- Tło
- Normalna fizjologia i patofizjologia potasu
- Potencjał czynnościowy kardiomiocytu bez rozrusznika
- Potencjał czynnościowy komórki rozrusznika serca
- Przewodzenie prądu
- Okres refrakcji
- Hyperkalemia, klasyfikacja i przyczyny
- Klasyfikacja
- Przyczyny
- Skutki hiperkaliemii
- Wpływ na metabolizm
- Pompa sodowo-potasowa
- Podsumowanie
Tło
Potas jest miękkim, srebrzystobiałym, wysoce reaktywnym kationem należącym do rodziny metali alkalicznych w układzie okresowym. Jest najobficiej występującym kationem w organizmie człowieka jako całości i najbardziej rozpowszechnionym jonem w jego przedziałach wewnątrzkomórkowych.
Średnio, zachodnia dieta zawiera od 80-100 mEq potasu dziennie, a w normalnych warunkach fizjologicznych, 90% z niego jest wchłaniane biernie, pozostawiając tylko 9,0 mmol do wydalenia z kałem. 3500-4000 mmol przechowywane w organizmie są nieproporcjonalne do dobowego poziomu potasu w osoczu, który jest normalnie utrzymywany w zakresie 3,5-5,3 mmol/L poprzez ścisły mechanizm homeostazy, przy czym najniższy poziom występuje w nocy i we wczesnych godzinach porannych, a najwyższy szczytowy poziom w godzinach popołudniowych.
Od momentu wchłonięcia do krwiobiegu, rolą nerek staje się dopasowanie spożycia potasu do jego wydatkowania; wymaga to kilku godzin, podczas których „wewnętrzna równowaga potasowa” pod wpływem insuliny i katecholamin utrzymuje tymczasową homeostazę poprzez przesuwanie potasu pomiędzy przestrzenią wewnątrzkomórkową i zewnątrzkomórkową. Stymulacja receptorów alfa upośledza wnikanie potasu do komórek, a stymulacja receptorów beta sprzyja temu w wyniku aktywacji pompy ATPazy sodowo-potasowej.
Pompa ATPazy sodowo-potasowej jest enzymem-ochroniarzem zlokalizowanym w sarkolemmie. Pomaga ona zabezpieczyć 98% potasu (około 144,0 mmol) zatrzymanego wewnątrz komórki. Zapewnia to zachowanie istotnej różnicy potencjałów przez błony komórkowe, potrzebnej do prawidłowego funkcjonowania komórek, zwłaszcza komórek pobudliwych, takich jak komórki nerwowe i komórki mięśnia sercowego.
Normalna fizjologia i patofizjologia potasu
Po jego szybkim wchłanianiu, potas pomaga orkiestrować własne poziomy ciała poprzez uwalnianie insuliny i aldosteronu. Inne nieodłączne bodźce organizmu, które również kontrolują poziom potasu w organizmie to receptory beta-2 adrenergiczne, zasadowe PH krwi i anabolizm komórkowy.
Uwolnienie insuliny i aldosteronu: Połknięty potas szybko dostaje się do krążenia. Po dotarciu do krążenia wrotnego, stymuluje trzustkę do uwalniania insuliny. Równocześnie krążący potas docierając do komórek kłębuszków nerkowych powoduje uwalnianie reniny. Renina po dotarciu do wątroby przekształca się w angiotensynę I. Angiotensyna I wędruje do płuc, gdzie przekształca się w angiotensynę II. Angiotensyna II następnie kończy swoją podróż z powrotem do nerek przez krążącą krew, aby pobudzić zona glomerulosa do wydzielania aldosteronu.
Wewnętrzna równowaga potasowa: Insulina uwolniona poposiłkowo działa przede wszystkim na mięśnie szkieletowe, aktywując dwa szlaki, szlak AKT-zależny odpowiedzialny za wstawianie transportera glukozy GLUT4 oraz szlak APK aktywujący komórkową ATPazę sodowo-potasową w celu przesunięcia potasu do przestrzeni wewnątrzkomórkowej. W przeciwieństwie do szlaku AKT-zależnego, szlak APK nie jest upośledzony ani przez zespół metaboliczny, ani przez przewlekłą chorobę nerek (ryc. 1).
Wydalanie: Potas filtrowany przez kłębuszki nerkowe jest biernie reabsorbowany w kanaliku proksymalnym i pętli Henlego proporcjonalnie do ilości dostarczonego sodu i wody. Normalnie tylko około 10% przefiltrowanego ładunku dociera do nefronu dystalnego.
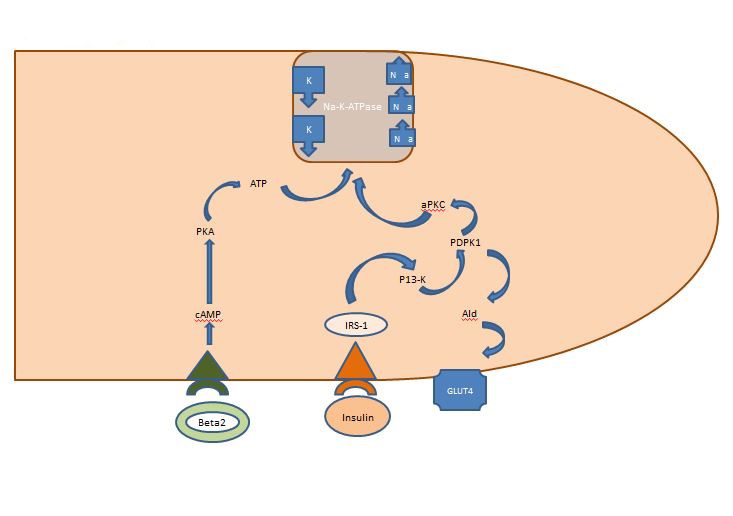
Rysunek 1. Działanie insuliny na komórkę mięśnia szkieletowego. Insulina uwalniana poposiłkowo aktywuje dwa szlaki w mięśniach szkieletowych, szlak AKT-zależny odpowiedzialny za przyłączenie transportera glukozy GLUT4 oraz szlak APK aktywujący komórkową ATPazę sodowo-potasową w celu przesunięcia potasu do przestrzeni wewnątrzkomórkowej.
Na początku kanalika dystalnego rozpoczyna się wydzielanie nadmiaru potasu, które stopniowo wzrasta w miarę posuwania się dalej w kierunku nefronu dystalnego i do kanalika zbiorczego. Jest to pośredniczone przez wzrost regulacji ATPazy wodorowo-potasowej na komórkach alfa-interkalacyjnych.
Obecność wyższych poziomów potasu w komórkach okołopęcherzykowych nerek aktywuje układ RAAS do uwalniania aldosteronu, który aktywuje ATPazę sodowo-potasową w błonie podstawnej, powodując spadek wewnątrzkomórkowego sodu, co prowadzi do zwiększenia transportu elektrogennego wychwytu potasu przez hiperpolaryzację napięcia błonowego i umożliwiając jego wydalanie z moczem.
W hiperkaliemii kontyngent potasu wydalanego przez jelito grube może wzrosnąć nawet o 30%, np, w przypadkach niewydolności nerek, gdzie potas jest następnie aktywnie pobierany przez aktywowaną pompę ATPazy sodowo-potasowej w błonie podstawnej enterocytów okrężnicy, aby zostać wydalony po drugiej stronie, do światła okrężnicy przez apikalne duże, zależne od wapnia kanały potasowe komórek.
Z powyższego wynika, że mechanizm homeostazy poziomu potasu w osoczu jest określony głównie przez interakcję trzech jednoczesnych transakcji – przyjmowania potasu, wewnątrz- i zewnątrzkomórkowych przesunięć potasu oraz wydalania potasu z moczem, z których wszystkie ostatecznie opierają się na pompie sodowo-potasowej.
Aby pojąć mechanizm nieuchronnego zagrożenia ze strony hiperkaliemii i jej zarządzania, należy zrozumieć fizjologię potencjału czynnościowego i wrodzone działanie enzymu ATPazy sodowo-potasowej.
Elektrofizjologia potencjału czynnościowego, tj, ruch jonów przez błony komórkowe, jest określony przez różnicę dwóch potencjałów, „potencjału chemicznego”, w którym jony poruszają się w dół ich gradientu stężenia i „potencjału elektrycznego”, w którym jony i cząsteczki odpychają podobne ładunki, dając potencjał transmembranowy (TMP), o którym mówi się, że jest +ve, gdy ruch netto jonów +ve jest na zewnątrz komórki i odwrotnie.
Potencjał czynnościowy kardiomiocytu bez rozrusznika
Potencjał czynnościowy składa się z pięciu faz, które rozpoczynają się i kończą w fazie 4. Pompy biorące udział w tym procesie to sarkolemma sodowo-wapniowa, ATPaza wapniowa i w końcu ATPaza sodowo-potasowa.
- Faza 4. Faza spoczynkowa: ma potencjał spoczynkowy wynoszący -90 mV w wyniku stałego ruchu potasu na zewnątrz przez kanały prostownika wewnętrznego. W tej fazie zamknięte są zarówno kanały sodowe, jak i wapniowe.
- Faza 0. Faza depolaryzacji: odpalenie komórki rozrusznikowej lub jej przewodzenie przez sąsiednią komórkę wyzwala wzrost TMP do wartości powyżej -90 mV. W tym momencie „szybkie kanały sodowe” zaczynają się otwierać jeden po drugim, pozwalając sodowi wniknąć do komórki, podnosząc TMP, a gdy wystarczająco dużo szybkich kanałów sodowych zostanie otwartych, aby uzyskać -70 mV, uruchomiony zostanie samopodtrzymujący się prąd sodowy, gwałtowna depolaryzacja TMP do 0 mV dla przejściowego okresu znanego jako „overshoot”, w którym to momencie zależne od czasu szybkie kanały sodowe zamykają się, a „długo otwierające się” kanały wapniowe otwierają się, aby podnieść TMP do -40 mV i umożliwić niewielki stały napływ wapnia w dół gradientu stężenia.
- Faza 1. Faza wczesnej repolaryzacji: rozpoczyna się od lekko +ve TMP i krótkotrwałego otwarcia niektórych kanałów potasowych, co powoduje jego przepływ na zewnątrz komórki, przywracając TMP z powrotem do około 0 mV.
- Faza 2. Faza plateau: tutaj dwa przeciwprądy są elektrycznie zrównoważone i skutkują utrzymaniem TMP w równowadze na poziomie nieco poniżej 0 mV. „Długo otwierające się kanały wapniowe” są nadal otwarte, co skutkuje stałym dopływem wapnia do komórki. Opóźniony prostowniczy kanał potasowy umożliwia przejście potasu na zewnątrz komórki w dół jej gradientu stężenia.
- Faza 3. Faza repolaryzacji: podczas tej fazy kanały wapniowe ulegają stopniowej inaktywacji, a utrzymujący się przepływ potasu na zewnątrz komórki przewyższa w ten sposób przepływ wapnia do wewnątrz, co powoduje powrót potasu do przestrzeni wewnątrzkomórkowej, a sodu i wapnia na zewnątrz komórki.
Potencjał czynnościowy komórki rozrusznika serca
Komórki rozrusznika serca mają wrodzony automatyzm, umożliwiający ich depolaryzację w rytmicznych cyklach. Węzeł zatokowo-przedsionkowy (SAN) ma najwyższy samoinicjowany rytm depolaryzacji z częstością 60-90/min, następnie węzeł przedsionkowo-komorowy (AVN) z częstością 40-60/min, a potem włókna Purkinjego i mięsień komorowy z częstością 20-40/min.
Potencjały błonowe komórek rozrusznikowych są niestabilne, a ich potencjały czynnościowe nie mają wyraźnych faz. Mają one mniej kanałów potasowych prostowników wewnętrznych, a ich TMP nigdy nie spada poniżej -60 mV, co eliminuje rolę szybkich kanałów sodowych, które wymagają TMP na poziomie -90 mV, co skutkuje brakiem fazy szybkiej depolaryzacji.
W TMP >-60 mV włącza się prąd „zabawowy/pacemakera” ze spontanicznym przepływem jonów przez wolne kanały sodowe, depolaryzując TMP do <-50 mV, a następnie z powrotem do -60 mV, gdy zamykają się kanały wapniowe.
Przewodzenie prądu
Wszystkie kardiomiocyty są elektrycznie sprzężone przez złącze szczelinowe, w tym komórka stymulatora. Ułatwia to rozległą depolaryzację wszystkich sąsiednich komórek, przekształcając serce w jedną funkcjonalną jednostkę, w której komórka o najwyższym współczynniku wrodzonym staje się „rozrusznikiem”.
Okres refrakcji
Dłuższy okres refrakcji podczas długiego plateau w fazie 2, spowodowany wolnymi kanałami wapniowymi, zapewnia czas potrzebny do całkowitego opróżnienia komór przed kolejnym skurczem. Okresy refrakcji mogą być bezwzględne (ARP), efektywne (ERP) lub względne (RRP). W ARP, komórka jest absolutnie uncitable.
An ERP trwa od ARP do krótkiego segmentu fazy 3. Bodziec w tym momencie może minimalnie depolaryzować komórkę, ale poziom depolaryzacji jest słabszy niż propagacja potencjału czynnościowego do sąsiednich komórek.
RRP jest wywołany przez ponadnormalny bodziec, prowadzący do depolaryzacji komórki i wytworzenia potencjału czynnościowego.
„Okres ponadnormalny” to stan nadpobudliwości, podczas którego słabszy niż normalny bodziec mógłby doprowadzić do arytmii, co wymusza synchronizację podczas kardiowersji w celu uniknięcia migotania komór (ryc. 2).
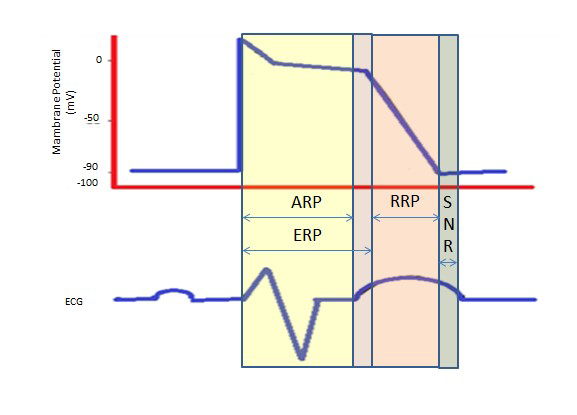
Ryc. 2. Okresy refrakcji. ARP: Absolute Refractory Period; ERP: Effective Refractory Period; RRP: Relative Refractory Period; SNR: Supranormal Refractory Period
Hyperkalemia, klasyfikacja i przyczyny
Klasyfikacja
Hyperkalemię klasyfikuje się jako łagodną, gdy poziomy są w zakresie 5,5-6,0 mmol/L, umiarkowaną od 6,1-6,9 mmol/L i ciężką przy poziomach 7.0 mmol/L lub większym, oraz przy każdym poziomie, przy którym występują zmiany w EKG .
Przyczyny
Hiperkaliemia występuje, gdy mechanizmy kompensacyjne nie są już w stanie poradzić sobie z zaburzeniem równowagi, dlatego zwykle jest wieloczynnikowa.
- Zwiększenie spożycia potasu jakąkolwiek drogą, np, doustne przyjmowanie pokarmu lub dożylne podawanie płynów zawierających potas, takich jak penicylina G.
- Zatrzymanie przez nerki: ponieważ wydalanie potasu zależy od aldosteronu i dostarczenia wystarczającej dystalnej ilości sodu i wody w obrębie nefronów, stany takie jak niewydolność nerek, niedoczynność nadnerczy (choroba Addisona) , hiporenemiczny hipoaldosteronizm typu IV, kwasica kanalików nerkowych, zwłaszcza u pacjentów z nefropatią cukrzycową, jak również każdy stan, który sprzyja hipoperfuzji, jak w przypadku zubożenia objętościowego i zastoinowej niewydolności serca, wpływają na skomplikowaną równowagę potasu w organizmie i predysponują do hiperkaliemii.
- Niedoczynność nadnerczy: musi być wykluczona u pacjentów z hiperkaliemią, szczególnie w obecności hiponatremii i osłabienia mięśni. W celu przesiewowego wykrycia pierwotnej niewydolności nadnerczy wykonuje się standardowy test stymulacji kosyntropiną, w którym podaje się 0,25 mg syntetycznej kosyntropiny w postaci bolusa dożylnego, po czym 45 minut do 1 godziny później dokonuje się pomiaru kortyzolu w osoczu. Wartości mniejsze niż 20 mcg/dL sugerują niewydolność nadnerczy.
- Leki zatrzymujące potas: leki na receptę, które zmniejszają aktywność ATPazy sodowo-potasowej, takie jak blokery receptorów beta-adrenergicznych, oraz leki zmniejszające wydzielanie aldosteronu, takie jak inhibitory ACE i ARB, niesteroidowe leki przeciwzapalne i leki moczopędne oszczędzające potas, wymagają ścisłej obserwacji w celu uniknięcia jatrogennej hiperkaliemii, zwłaszcza w geriatrycznej grupie wiekowej z postępującym spadkiem czynności nerek jako części procesu starzenia.
- Zaburzenia w transkomórkowym przemieszczaniu potasu: mogą wystąpić w warunkach kwasicy, hiperglikemii, hiperosmolalności, ciężkiego wysiłku fizycznego, rozpadu tkanek, okresowego porażenia hiperkaliemicznego oraz podczas stosowania leków blokujących receptory beta-adrenergiczne. Na każde zmniejszenie PH krwi o 0,1 jednostki, stężenie potasu w surowicy wzrasta o około 0,6 mmol/L (mniej, jeśli kwasica jest spowodowana przez kwasy organiczne) .
- Pseudo-hypoaldosteronizm jest wrodzoną chorobą dziedziczoną autosomalnie recesywnie, w której nerki są oporne na działanie aldosteronu.
- Pseudohiperkaliemia również nie może być przeoczona: jak sama nazwa wskazuje, występuje wtedy, gdy jest podwyższony potas w surowicy w obecności prawidłowego potasu w osoczu. Można ją zaobserwować przy hemolizie krwi, długotrwałym zaciskaniu opaski uciskowej podczas procedury pobierania krwi, co powoduje zewnątrzkomórkowe uwalnianie potasu, przy powtarzającym się zaciskaniu pięści podczas flebotomii, traumatycznym nakłuwaniu żył, przy leukocytozie i trombocytozie oraz w niektórych rzadkich zespołach genetycznych, takich jak rodzinna pseudohiperkaliemia i dziedziczna sferocytoza. Może to być jednak po prostu wynik zwykłego błędu laboratoryjnego.
Skutki hiperkaliemii
Łagodna hiperkaliemia jest często bezobjawowa, wykrywana przypadkowo w badaniach laboratoryjnych, ze względu na niejasne objawy, takie jak złe samopoczucie, osłabienie mięśni i parestezje. Ciężka hiperkaliemia wpływa na funkcje nerwowo-mięśniowe w postaci osłabienia i porażenia mięśni szkieletowych; nie jest to jednak częsty objaw, ponieważ toksyczność sercowa dominuje w obrazie i jest objawem wstępnym. Toksyczność sercowa będzie zazwyczaj występować w EKG w następujący, narastający sposób, chociaż niekoniecznie, w zależności od etiologii:
- Przy stężeniach większych niż 5,5 mEq/L, wzrost przewodnictwa kanałów potasowych zwiększa prąd lkr, prowadząc do szybkiej repolaryzacji w postaci szczytowej fali T w powierzchniowym EKG. Te fale T można odróżnić od fal występujących w zawale mięśnia sercowego i CVA dzięki krótkiemu czasowi trwania, wynoszącemu 150-250 msec.
- Przy stężeniu potasu większym niż 6,5 mEq/L występuje stan utrzymującej się depolaryzacji podprogowej, powodujący opóźnienie depolaryzacji przedsionków i komór. Zmniejszenie fazy 0 potencjału czynnościowego prowadzi do wydłużenia potencjału czynnościowego, powodując opóźnienie przewodzenia śródkomorowego i przedsionkowo-komorowego. W powierzchniowym zapisie EKG objawia się to spłaszczeniem i zanikiem załamków P oraz poszerzeniem zespołów QRS. Wraz z narastającym opóźnieniem przewodzenia śródkomorowego, w powierzchniowym EKG zaczynają pojawiać się objawy bloku lewej i prawej odnogi pęczka Hisa. Można go odróżnić od choroby odnogi pęczka Hisa dzięki temu, że w hiperkaliemii opóźnienie utrzymuje się przez cały czas trwania zespołu QRS, a nie tylko odpowiednio w początkowej lub końcowej jego części.
- Przy stężeniu 10 mEq/L przestaje występować przewodzenie zatokowo-przedsionkowe, a rolę tę przejmuje przyspieszony rytm węzłowy. Komorowe zaburzenia rytmu rozwijają się wraz z łączeniem się poszerzonych zespołów QRS z falami T, które ostatecznie tworzą klasyczny wzór fali sinusoidalnej. Gdy to nastąpi, VF i asystolia są nieuchronne, a następnie dochodzi do zatrzymania krążenia.
- Czasami zmiany mogą być nieregularne i nieprzewidywalne, a EKG będzie przeskakiwał od normy do asystolii z powodu zmienności czynników etiologicznych i ich wpływu, np. szybkości zmian potasu, stężenia wapnia, pH i stężenia sodu. Dlatego też hiperkaliemia powinna być leczona w trybie pilnym, gdy stężenie potasu przekracza 6,5 mmol/l lub w przypadku wystąpienia objawów hiperkaliemii w EKG niezależnie od stężenia potasu. Inne zgłaszane związki z ostrą hiperkaliemią to: obraz pseudo MI w zapisie EKG, z masywnym odcinkiem ST-T w wyniku zaburzeń repolaryzacji miocytów, krótki odstęp PR i QT, tachykardia zatokowa, bradykardia zatokowa, rytm idiowentrykularny, blok serca I i II stopnia.
Wpływ na metabolizm
Hiperkaliemia prowadzi do hiperchloremicznej kwasicy metabolicznej, ponieważ hiperkaliemia promuje wewnątrzkomórkowy wychwyt potasu w zamian za jony wodorowe. Powoduje to alkalozę wewnątrzkomórkową, hamując wytwarzanie amoniaku przez nerki w kanalikach proksymalnych, co prowadzi do zmniejszenia wydalania amonu i kwasu z moczem oraz kwasicy kanalikowej typu IV.
Pompa sodowo-potasowa
Atpaza sodowo-potasowa została odkryta w 1957 roku przez Skou, który później otrzymał za swoje odkrycie część Nagrody Nobla z chemii w 1997 roku.
Skou jako pierwszy odkrył ATPazę sodowo-potasową w sarkolemmie powierzchni komórek mięśni sercowych. Jej obecność została później wykryta w każdym eukariotycznym jedno- i wielokomórkowym organizmie.
Pompa sodowo-potasowa funkcjonuje poprzez łączenie hydrolizy ATP z komórkowym eksportem trzech jonów sodu w zamian za dwa jony potasu wbrew ich gradientom elektrochemicznym. Jest ona celem molekularnym naparstnicy i digoksyny, które są stosowane od XVIII wieku jako wyciągi z naparstnicy.
Działanie pompy sodowo-potasowej jest regulowane przez fosfoproteinowy fosfolemman, którego niefosforylacja prowadzi do zahamowania pompy, a fosforylacja prowadzi do zwiększenia aktywności pompy. Posiada on trzy miejsca fosforylacji, dwa miejsca palmitoilacji i jedno miejsce glutationylacji, co tłumaczy mnogość sygnałów zdolnych do pobudzania i hamowania pompy.
Sama pompa sodowo-potasowa jest enzymem złożonym z wielu podjednostek o wielu izoformach. Obecność podjednostek alfa i beta (głównie B1 w sercu) jest niezbędna do jej funkcjonowania. Ostatnio w nerkach zidentyfikowano trzecią podjednostkę białka gamma, ale do tej pory jej funkcja pozostaje nieznana.
Podjednostka alfa stanowi katalityczny rdzeń enzymu pompy sodowo-potasowej. Ma około 100 kDa i zawiera miejsca wiążące dla sodu, potasu, ATP i steroidów kardiotonicznych, takich jak ouabaina. Tylko alfa 1 i alfa 2 wykazują znaczącą obecność w prawidłowym miocycie serca i są funkcjonalnie związane z sodowo-wapniowym wymiennikiem (NCX). Alfa 3 została zgłoszona do zastąpienia alfa 2 w eksperymentalnych modelach niewydolności serca .
Dane z ostatnich eksperymentów sprzyjają udziałowi obu podjednostek alfa 1 alfa 2 pompy w regulacji sprzężenia pobudzenie-skurcz (E-C). Uważa się, że alfa 1, która została znaleziona, aby być bardziej równomiernie rozmieszczona w całej sarkolemmie, odgrywa więcej roli „porządkowej”, kontrolując zarówno kurczliwość, jak i luzem wewnątrzkomórkowy sód, podczas gdy alfa 2, której ekspresja jest skoncentrowana w T-tubulach wraz z innymi kluczowymi składnikami sprzężenia E-C, koncentruje się głównie na kurczliwości .
Znane czynniki, które mogą kontrolować pompę sodowo-potasową, obejmują: ATP, sód wewnątrzkomórkowy, bariery podsurowicówkowe i przestrzenie rozmyte, potencjał błonowy, wewnątrzkomórkowe szlaki sygnalizacyjne (adrenergiczne szlaki sygnalizacyjne, kinaza białkowa A & C, tlenek azotu, fosfolemana), bezpośrednia regulacja przez małe cząsteczki (lipidy, endogenne steroidy kardiotoniczne), inne białka stowarzyszone (caveolae i caveoliny oraz ankyrina).
Podsumowanie
Hiperkaliemia jest wyzwaniem klinicznym i może występować nawet u 10% hospitalizowanych pacjentów. Jej końcowy efekt jest groźny dla życia. Ponieważ na wszystkie komórki organizmu ostatecznie wpływa pompa sodowo-potasowa, a wiadomo, że niedokrwione mięśnie sercowe wydzielają potas pozakomórkowo, co prowadzi do obniżenia progu arytmii z możliwością wystąpienia arytmii komorowych, które nasilają hipopolaryzację i jeszcze bardziej obniżają próg, należy skoncentrować więcej badań na manipulacji enzymem sodowo-potasowym, ponieważ jego kontrola mogłaby korzystnie zmienić wyniki zatrzymania krążenia i przeformułować aktualne wytyczne resuscytacji.