ABOVE: modified from © ISTOCK.com, tera vector
Prawie zawsze zbudowanie czegoś jest trudniejsze niż zburzenie tego. Podobnie, wbijanie genów stanowi większe wyzwanie niż ich wybijanie. Jest to rzeczywistość, z którą naukowcy będą musieli się zmierzyć, aby uzyskać jak najwięcej korzyści z edycji genów. Wbijanie genów pozwala naukowcom badać efekty konkretnych wariantów genów, wykorzystywać geny reporterowe, takie jak białko zielonej fluorescencji, do śledzenia produktów genowych w czasie i przestrzeni, do badania regulacji genomu, a w końcu do naprawy genów wywołujących choroby. „To naprawdę skuteczny sposób na zbadanie każdej bazy w genie” – mówi Greg Findlay, doktorant na Uniwersytecie Waszyngtońskim.
CRISPR-Cas9, technologia edycji genów znana z łatwości obsługi, może wprowadzać lub usuwać geny. Znokautowanie genu polega na wprowadzeniu CRISPR-Cas9 do komórki przy użyciu RNA przewodnika, który kieruje narzędzie do interesującego nas genu. Tam Cas9 przecina gen, przecinając obie nici DNA, a regularny mechanizm naprawy DNA komórki naprawia przecięcie za pomocą procesu zwanego niehomologicznym łączeniem końców (NHEJ). NHEJ jest bardzo wydajny, ale niedokładny. Proces ten ma tendencję do wprowadzania błędów w postaci małych wstawek lub delecji, które zwykle wystarczają do zlikwidowania genu.
Aby jednak wprowadzić gen, cięcia muszą być naprawione bardzo precyzyjnie, bez dodatkowych wstawek lub delecji. Wymaga to wykorzystania drugiego mechanizmu naprawy DNA, zwanego naprawą ukierunkowaną na homologię (HDR), który – przynajmniej w komórkach ssaków – zachodzi mniej wydajnie, więc jego częstotliwość jest mniejsza niż NHEJ. Proces ten dodatkowo komplikuje fakt, że niektóre loci genowe i typy komórek są z natury mniej podatne na edycję CRISPR-Cas9.
W ciągu ostatnich kilku lat naukowcy opracowali wiele nowych strategii, aby zwiększyć wydajność pukania w geny zarówno duże, jak i małe za pomocą CRISPR-Cas9, a po drodze zaproponowali i przetestowali nowe zastosowania dla tego typu edycji genów. Tutaj The Scientist analizuje kilka najbardziej obiecujących podejść.
Select It
Badacz: Jon Chesnut, starszy dyrektor ds. biologii syntetycznej R&D, Thermo Fisher Scientific
Projekt: Opracowując zestaw do znakowania genów o nazwie Truetag, który firma Thermo Fisher wprowadzi na rynek jeszcze w tym roku, Chesnut wykorzystał markery selektywne do poprawy
wydajności. Selektywny marker – w tym przypadku gen oporności na antybiotyki – jest przyklejany do znacznika białka fluorescencyjnego i wprowadzany do komórek ssaków. Komórki te są następnie hodowane w kulturze z powiązanym antybiotykiem. Gen oporności daje przewagę selektywną komórkom, które są jego nosicielami; tylko one są w stanie rosnąć, a zatem te, które rosną, zawierają znacznik interesującego nas genu. Nawet jeśli wydajność wstawiania genów jest niska, badacze mogą stosować selekcję antybiotykową przez tydzień lub dłużej, aby uzyskać wysoki procent komórek z udanymi wstawkami.
Używając antybiotyku puromycyny lub blasticidyny z zestawem, zespół Chesnuta zdołał zwiększyć szybkość wstawiania genów z 10-30 procent do 90 procent lub więcej w niektórych populacjach komórek. Kilka szczególnie trudnych genów przeszło z poziomu mniejszego niż 1 procent do wyższego niż 90 procent. Ważne jest, aby przetestować wiele dawek antybiotyków na linii komórkowej, którą planujesz użyć, aby znaleźć właściwą dawkę, mówi Chesnut: chcesz zabić komórki bez wstawek, ale nie komórki z udanymi wstawkami.
Spróbuj: Markery selektywne działają najlepiej, gdy interesujący nas gen ma wysoką ekspresję, mówi Chesnut. „Jeśli nie jest, nadal możesz uzyskać selekcję, ale możesz nie uzyskać wystarczającej ekspresji znacznika białka fluorescencyjnego, aby być w stanie go wykryć”. Również ogólne ograniczenia CRISPR-Cas9 mają zastosowanie. „Istnieją regiony genomu, które nie tną się bardzo dobrze z CRISPR, i wciąż nie jesteśmy pewni dlaczego” – dodaje. A niektóre typy komórek nie akceptują łatwo obcego DNA, RNA lub kompleksów RNA-białko – trzech metod dostarczania CRISPR-Cas9.
Aby uzyskać więcej szczęścia przy wstawianiu wybieralnych markerów, należy upewnić się, że istnieje tak zwana sekwencja PAM, krótki znacznik w docelowym DNA, który CRISPR-Cas9 musi rozpoznać, zanim dokona cięcia, w obrębie 10 par zasad od pożądanego miejsca wstawienia genu, mówi Chesnut. Dalsza odległość od miejsca cięcia może spowodować, że wydajność wstawiania będzie zbyt niska, aby było to funkcjonalne. Bez miejsca PAM można spróbować TALEN-ów lub nukleaz z palcem cynkowym, choć te starsze techniki edycji genów są trudniejsze niż CRISPR.
Timed Inhibition
Badacz: Jacob Corn, biolog genomu, Swiss Federal Institute of Technology, Zurich
Projekt: Naukowcy nie rozumieją, dlaczego szlak NHEJ znacznie przewyższa szlak HDR w komórkach ssaków. „Drożdże wykonują HDR jak szalone,” mówi Corn. Aby ożywić ten proces naprawy DNA w ludzkich komórkach i poprawić kontrolę nad wbijaniem genów, Corn i jego zespół próbują ustalić, jak regulowany jest HDR. Przebadali ludzkie komórki w poszukiwaniu genów, których knockdown prowadził do zwiększenia HDR w komórce, a następnie szukali małych molekuł hamujących te geny. Jeden z genów, który się pojawił, koduje CDC7, kinazę, która reguluje przejście cyklu komórkowego do fazy S; jej inhibitor, XL413, zwiększył wydajność knock-in genu dwu- lub trzykrotnie (BioRXiv, DOI: 10.1101/500462, 2018). To dlatego, że HDR występuje tylko w niektórych częściach cyklu komórkowego, w tym w fazie S, mówi Corn. Jeśli dodasz inhibitor XL413 w tym samym czasie, gdy używasz CRISPR-Cas9 do edycji docelowego genu, komórki piętrzą się w fazie bezpośrednio przed fazą S. Następnie usuwasz XL413, a wszystkie komórki przechodzą do fazy S i zwiększają wydajność knock-in.

Spróbuj: „Czas jest absolutnie kluczowy,” mówi Corn. Cas9 musi przeciąć DNA w tym samym czasie, w którym dodawany jest XL413. Jeśli najpierw zahamujesz, a potem uwolnisz podczas edycji za pomocą CRISPR-Cas9, wydajność rekombinacji homologicznej spadnie trzykrotnie, zamiast wzrosnąć, ponieważ komórki zostaną uwolnione do niewłaściwej fazy cyklu komórkowego.
I tak jak w przypadku każdego wysiłku HDR, Corn mówi, zawsze przeprowadzaj kontrolę bez nukleazy, aby upewnić się, że nie amplifikujesz przypadkowo zanieczyszczonego DNA, które krąży po laboratorium. Po wprowadzeniu knock-in, „sekwencja, sekwencja, sekwencja, sekwencja”, mówi. Samo użycie systemu reporterowego, takiego jak znacznik białka fluorescencyjnego, aby zademonstrować udaną insercję genu może okazać się nieskuteczne. Sekwencjonowanie weryfikuje, czy wstawki zostały wykonane w odpowiednim miejscu.
Playing the Long Game
Researcher: Channabasavaiah Gurumurthy, dyrektor placówki zajmującej się inżynierią genomu myszy, University of Nebraska Medical Center
Projekt: Kilka lat temu, zastanawiając się nad trudnościami związanymi z wprowadzaniem genów do zygot myszy, Gurumurthy i jego koledzy mieli objawienie.
Badacze z powodzeniem wstawiali krótkie, jednoniciowe DNA, więc dlaczego nie spróbować zrobić knock-in przez wstawianie długich, jednoniciowych DNA? Rzeczywiście, podejście, które Gurumurthy nazywa Easi-CRISPR (wydajne dodatki z wstawkami ssDNA -CRISPR), zwiększa wydajność o 2,5 raza, a użycie jednoniciowego DNA tnie wskaźnik wstawek poza celem 100-krotnie w kulturze komórkowej (Nat Protoc 13:195-215, 2018; Nature 559:405-09, 2018). „To jest dość ogromne” – mówi. W laboratorium Gurumurthy’ego, Easi-CRISPR wygenerował linię myszy knock-in dla 9 z każdych 10 genów, które wypróbowali. Współpracownik wykorzystał ją również w ludzkich limfocytach T do stworzenia komórek CAR-T, komórek odpornościowych specyficznych dla pacjenta, przeznaczonych do walki z rakiem.
Spróbuj: Easi-CRISPR jest daleki od niezawodności, ostrzega Gurumurthy. Czasami technika ta wstawia tylko część genu. Ponadto, dodaje, może ona zakłócić ramiona homologii – krótkie sekwencje po obu stronach genu, które kierują go do właściwego celu w genomie. A niektóre loci są niewytłumaczalnie trudniejsze do wstawienia niż inne.
Niewielu komercyjnych sprzedawców projektuje i syntetyzuje niestandardowe długie, jednoniciowe DNA. Można stworzyć własne, ale stabilność jednoniciowego DNA jest różna; mniej stabilne sekwencje będą miały niższą wydajność, więc może być konieczne zsyntetyzowanie ich większej ilości, mówi Gurumurthy.
Badacze, którzy nie są w stanie wstawić CRISPR do jednokomórkowych embrionów myszy, mogą zapłacić ośrodkowi za stworzenie myszy z ich sekwencją DNA, mówi Gurumurthy. Ośrodki takie jak jego pobierają opłaty w wysokości od 5 000 do 15 000 USD za wygenerowanie jednej lub dwóch par hodowlanych; ośrodki komercyjne pobierają opłaty w wysokości 20 000-50 000 USD, mówi.
Knock-in By Numbers
Badacz: Greg Findlay, kandydat na MD/PhD w laboratorium Jaya Shendure’a, University of Washington
Projekt: Findlay i jego koledzy mieli na celu poprawę sposobu, w jaki lekarze klinicyści interpretują mutacje w genie BRCA1 odpowiedzialnym za raka piersi i jajników. Gen ten ma tysiące wariantów, ale badacze nie wiedzą, jak większość z nich wpływa na jego działanie. Aby zbadać wpływ tych wariantów, wykorzystali opracowaną przez siebie technikę knock-in zwaną nasyconą edycją genomu (Nature, 562:217-22, 2018).
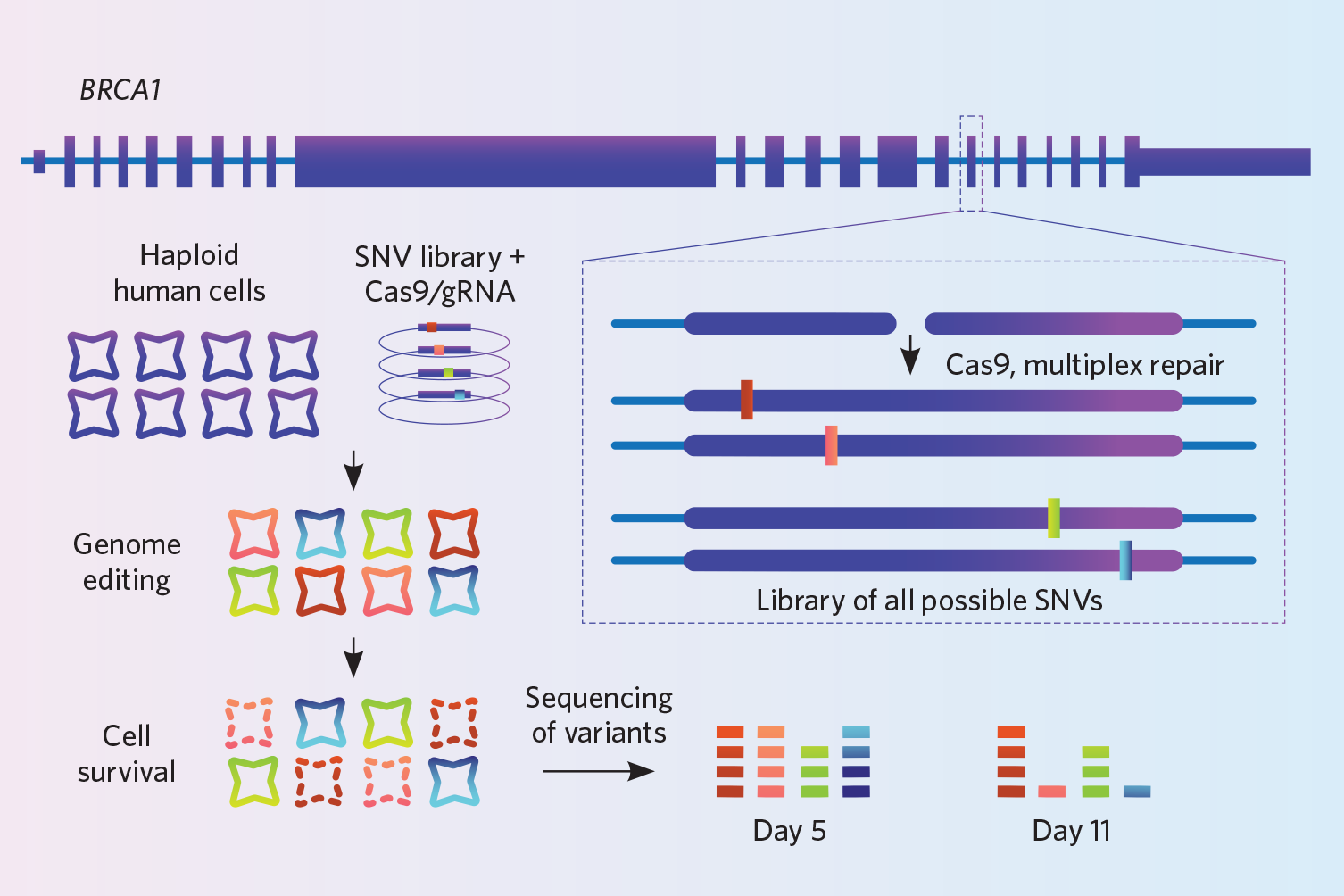
W unieśmiertelnionej haploidalnej ludzkiej linii komórkowej, użyli CRISPR-Cas9, aby wbić 4000 drobnych wariantów w milionach komórek jednocześnie in vitro. Genom jest cięty w tym samym miejscu w każdej komórce, ale genom każdej komórki otrzymuje inny wariant. Aby promować HDR, znokautowano również gen ligazy4, wyłączając ścieżkę naprawczą NHEJ – krok, który przyniósł trzykrotny wzrost wydajności, mówi Findlay. Wreszcie, ponieważ wszystkie komórki są różne, sekwencjonowali komórki dogłębnie, obejmując ten sam region genomu miliony razy, aby upewnić się, że rzeczywiście znokautowali 4,000 wariantów, które chcieli zbadać. Sekwencjonowali w dwóch punktach czasowych i wywnioskowali, że knock-ins, które nie pojawiły się w sekwencjonowaniu w drugim punkcie czasowym były tymi, które zakłócały funkcję genu, ponieważ komórki je niosące musiały umrzeć.
Spróbuj: Zespół Findlay’a miał oligo DNA dla 4,000 wariantów wyprodukowanych dla nich na mikromacierzy. Można kupić tablice zawierające od 6 000 do 250 000 oligosów, więc należy rozważyć uzyskanie większych korzyści poprzez połączenie wielu eksperymentów na tej samej tablicy, mówi Findlay. Ich laboratorium płaci około 5 000 dolarów za 100 000 oligosów.
Strategia ta ma pewne ograniczenia: jak dotąd była stosowana tylko do wprowadzania wariantów pojedynczych nukleotydów, a wszystkie zmiany muszą dotyczyć tego samego genu. Metoda ta działa najlepiej, gdy edytuje się dość wąski region DNA, około 110-120 par zasad, ponieważ dłuższe oligo DNA zawierałyby zbyt wiele błędów, mówi Findlay. Ważne jest również, aby sekwencjonować bardzo głęboko, aby upewnić się, że uwzględnia się pełną liczbę wariantów, które zamierzamy wprowadzić.