- A Clinicopathologic Review
- INTRODUCTION
- EPITELIALNO-STROMALNE DYSTROFIE ROGÓWKI
- DYSTROFIA ROGÓWKI REIS-BUCKLERSA
- LATTICE CORNEAL DYSTROPHY
- GRANULARNA DYSTROPIA ROGÓWKI, TYP I
- GRANULAR CORNEAL DYSTROPHY, TYPE II
- STROMALNA DYSTROFIA ROGÓWKI
- MACULARNA DYSTROFIA ROGÓWKI
- DYSTROPIA ROGÓWKI SCHNYDERA (SCD)
- Tabela 4: Mnemonik do zapamiętania dystrofii zrębu rogówki
- OVERVIEW: CORNEAL STROMAL DYSTROPHIES
- EPIDEMIOLOGIA
- ZNACZENIA
- SYMPTOMY
- Postępowanie
A Clinicopathologic Review
Emily S. Birkholz, MD, Nasreen A. Syed, MD, and Michael D. Wagoner, MD, PhD
August 17, 2009
Major Revision: Chaunhi Van, MD and Nasreen Syed, MD
August 20, 2015
INTRODUCTION
Dystrofie nabłonkowo-stromalne i stromalne rogówki są grupą dziedzicznych zaburzeń rogówki, które są spowodowane postępującym gromadzeniem się złogów w obrębie warstw rogówki. Złogi te nie są spowodowane zapaleniem, infekcją lub urazem, ale mutacjami genetycznymi, które prowadzą do transkrypcji nieprawidłowych białek, co skutkuje gromadzeniem się nierozpuszczalnego materiału w obrębie rogówki. Zaburzenia te mogą, ale nie muszą wpływać na widzenie i mogą, ale nie muszą być symetryczne (1). System klasyfikacyjny 2015 International Committee for Classification of Corneal Dystrophies (IC3D) podzielił dystrofie rogówki na 4 kategorie: dystrofie nabłonkowe i podnabłonkowe, dystrofie nabłonkowo-zrębowe, dystrofie stromalne oraz dystrofie śródbłonkowe. Większość dystrofii wcześniej uznawanych za dystrofie stromalne jest obecnie klasyfikowana jako dystrofie nabłonkowo-stromalne lub dystrofie stromalne. Tabela 1 i 2 zawierają listę dystrofii nabłonkowo-stromalnych i dystrofii stromalnych (2). Stara klasyfikacja dystrofii stromalnych rogówki jest wymieniona w tabeli 3.
- Dystrofia rogówki Reisa-Bucklersa
- Dystrofia rogówki Thiela-Behnke
- Dystrofia rogówki typu Lattice, typ 1 i warianty
- Ziarnista dystrofia rogówki, typ 1
- Ziarnista dystrofia rogówki, typ 2
- Macularna dystrofia rogówki
- Dystrofia rogówki Schnydera
- Wrodzona stromalna dystrofia rogówki
- Tylna bezpostaciowa dystrofia rogówki
- Centralna mętna dystrofia Francois
- Przed- i po-.Descemet corneal dystrophy
Tabela 3. Stara klasyfikacja dystrofii zrębu rogówki
- Lattice corneal dystrophy
- Granular corneal dystrophy
- Avellino corneal dystrophy
- Macular corneal dystrophy
- Gelatinous drop-jak dystrofia
- Dystrofia rogówki Schnydera
- Dystrofia rogówki Francois-Neetans Fleck dystrofia
- Wrodzona dziedziczna dystrofia stromalna
EPITELIALNO-STROMALNE DYSTROFIE ROGÓWKI
Dystrofie epitelialno-stromalne są spowodowane mutacjami w genie transformującego czynnika wzrostu beta (TGFβI), znanego również jako gen BIGH3. TGFβI jest zlokalizowany na chromosomie 5q31 i koduje keratoepitelinę, białko wydzielane przez nabłonek rogówki. Białko to działa jako białko adhezyjne i jest obecne w prawidłowym zrębie. Będąc małym białkiem o wielkości zbliżonej do albuminy, ma zdolność dyfuzji przez zrąb rogówki. W przypadku mutacji w genie TGFβI struktura keratoepiteliny jest nieprawidłowa i dochodzi do akumulacji nierozpuszczalnego białka lub jego proteolitycznych fragmentów w rogówce (1, 3). Co ciekawe, mutacja genu TGFβI została odkryta częściowo na Uniwersytecie w Iowa. Grupa badaczy i klinicystów, w tym Edwin M. Stone, Robert Folberg i Jay H. Krachmer, w 1994 roku przypisała dystrofię ziarnistą typu I, ziarnistą typu II i dystrofię siatkowatą do chromosomu 5q (4). Do chwili obecnej zidentyfikowano 63 różne mutacje w genie TGFβI. Nie zidentyfikowano skutecznych metod leczenia zapobiegających lub zmniejszających odkładanie się keratoepiteliny. Dystrofie mają zwykle dziedziczenie autosomalne dominujące i dotyczą warstwy Bowmana oraz zrębu (3).
DYSTROFIA ROGÓWKI REIS-BUCKLERSA
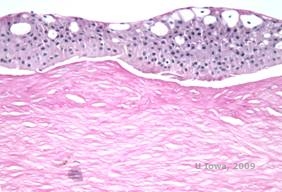
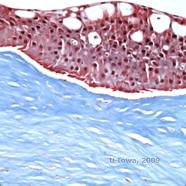
Dystrofia rogówki Reisa-Bücklersa, znana wcześniej jako ziarnista dystrofia rogówki typu III lub dystrofia rogówki Bowmana typu I, zwykle charakteryzuje się prawidłową rogówką po urodzeniu, ale w ciągu pierwszej dekady życia rozwijają się bolesne, nawracające nadżerki, opakeracja i postępująca utrata wzroku (1). W warstwie Bowmana i zrębie przednim zlokalizowane są nieregularne, szarobiałe, geograficznie podobne zmętnienia. W bardziej zaawansowanych stadiach choroby, zmętnienia mogą rozciągać się na limbus i głębsze warstwy zrębu (2). Badanie histopatologiczne ujawnia przednie zręby i podnabłonkowe złogi materiału podobnego do hialiny, które przerywają i często zastępują warstwę Bowmana (patrz rysunek 1A i 1B). Złogi te barwią się na czerwono barwnikiem trichromowym Massona (2). Materiał hialinopodobny składa się z prętopodobnych ciał ultrastrukturalnych, co pozwala odróżnić tę chorobę od dystrofii rogówki Thiel-Behnke (1, 2).
| A: H&E Reisa Bücklera pokazujące zniszczenie warstwy Bowmana i nieregularny nabłonek | B. Masson Trichrome stain demonstrating epithelial staining |
 |
 |
LATTICE CORNEAL DYSTROPHY
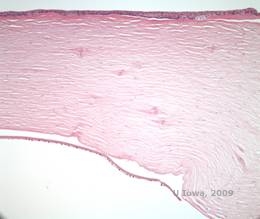
Lattice corneal dystrophy (LCD) is the most common of the corneal epithelial-stromal dystrophies. Jest to choroba obustronna, dziedziczona autosomalnie dominująco, która zwykle ujawnia się pod koniec pierwszej dekady życia z objawami nawracających nadżerek rogówki i pogorszenia widzenia. Charakteryzuje się liniami siatkowatymi, które są liniowymi, promieniście zorientowanymi, rozgałęziającymi się refrakcyjnymi zmętnieniami opisywanymi jako „podobne do szkła”, zlokalizowanymi w zrębie przednim (patrz rysunek 2A i 2B). Linie siatki znajdują się początkowo w powierzchownej, centralnej części rogówki. W miarę postępu choroby rozprzestrzeniają się one głębiej i obwodowo w zrębie, z pominięciem limbusu (1, 2). Inne wyniki badania obejmują plamiste nieprzezroczystości, podnabłonkowe białe kropki oraz „szkliste” zamglenia zrębu, które zaczynają się centralnie i stają się bardziej rozproszone (2). Wielu pacjentów z LCD będzie wymagało interwencji chirurgicznej w celu leczenia nawracających nadżerek i pogorszenia widzenia. Jeżeli choroba zlokalizowana jest w przedniej części zrębu, pacjenci często mogą być skutecznie leczeni za pomocą keratektomii fototerapeutycznej (PTK). Niektórzy wymagają przeszczepu rogówki. Ponieważ keratoepitelina, białko produkowane przez gen TGFβI, jest wytwarzane głównie w nabłonku rogówki, choroba ma tendencję do nawrotów w przeszczepach rogówki (1).
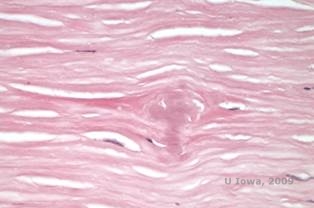
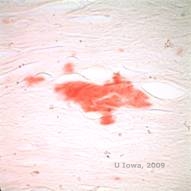
W LCD złogi amyloidu gromadzą się pomiędzy błoną podstawną nabłonka i warstwą Bowmana, jak również w zrębie, powodując zniekształcenie architektury blaszki. Złogi te barwią się pozytywnie w badaniu immunohistochemicznym z użyciem przeciwciał przeciwko keratoepitelinie (2). Złogi pojawiają się jako bezpostaciowe różowe złogi w barwieniu hematoksyliną i eozyną (H&E) (patrz rysunek 1C i 1D) i barwią się czerwienią Kongo, wykazując klasyczną zieloną dwójłomność w polaryzacji krzyżowej (patrz rysunek 2E i 2F) (1). Brak lub ścieńczenie warstwy Bowmana, atrofia nabłonka i degeneracja nabłonka podstawnego mogą być również stwierdzone w badaniu histopatologicznym w LCD (2).
LCD typu I jest klasyczną postacią LCD spowodowaną mutacją w genie TGFβI, której skutkiem jest izolowane odkładanie amyloidu w rogówce. Zidentyfikowano cztery warianty LCD: LCD typu IIIA, typu I/IIIA, typu IV oraz amyloidozę polimorficzną. Warianty LCD pojawiają się w późniejszym okresie życia niż klasyczne LCD. LCD typu IIIA pojawia się w 5-7 dekadzie życia, zwykle z nadżerkami nabłonka. Charakteryzuje się grubszymi liniami siatkowatymi, opisywanymi jako „wyglądające jak ropa”, które rozciągają się aż do limbusu. LCD typu I/IIIA charakteryzuje się cienkimi liniami siatkowatymi. LCD typu IV pojawia się w 7-9 dekadzie życia z małymi liniami siatkowatymi. Złogi amyloidu w LCD typu IV znajdują się w głębokim zrębie i rzadko dochodzi do nadżerek nabłonka. W polimorficznym typie amyloidozy linie siatkowate są nieobecne i rzadko dochodzi do nadżerek nabłonka (2).
LCD typu II jest układowym zespołem amyloidozy znanym jako zespół Meretoja, dotyczącym skóry, nerwów czaszkowych i rogówki. Występuje we wczesnej dorosłości z neuropatią obwodową, neuropatią czaszkową, twarzą przypominającą psa gończego, suchością skóry, blepharochalasis, wystającymi wargami i liniami siatki rogówki. Ten typ został powiązany z genem gelsoliny na chromosomie 9, który koduje białko prekursorowe amyloidu, które funkcjonuje w celu usunięcia aktyny z miejsc urazu i zapalenia (1). Nazwa ta jest błędna i nie jest uważana za wariant dystrofii rogówki typu lattice (2).
| A: Lewe oko w retroiluminacji demonstrujące przednie złogi zrębu w lattice corneal dystrophy | B: Lewe oko z wyższą mocą pokazujące liniowe przednie złogi zrębu. |
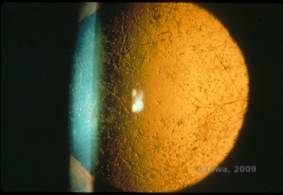 |
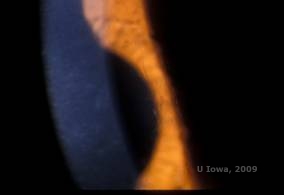 |
| C: Barwienie H&E rogówki z łaciatością. Uwaga na różowe, amorficzne złogi w zrębie | D: Bliższy widok różowych, amorficznych złogów |
 |
 |
| E: Barwienie czerwienią Kongo, podkreślenie amyloidu | F: Jabłkowo-zielona dwójłomność amyloidu z polaryzacją poprzeczną. |
 |
 |
GRANULARNA DYSTROPIA ROGÓWKI, TYP I
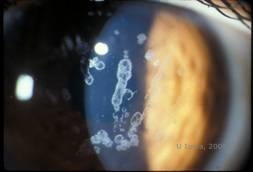
Granularna dystrofia rogówki, typu I (GCD1) jest obustronną, dziedziczoną autosomalnie dominująco chorobą związaną z mutacją w genie TGFβI, która prowadzi do odkładania się hialinowego materiału w zrębie rogówki. Typowym objawem choroby jest pojawienie się na początku pierwszej dekady życia szaro-białych, „okruchowych” zmian w zrębie przednim i środkowym, rozszerzających się na zrąb tylny w zaawansowanym stadium choroby (1, 2). Zmętnienia te są dyskretnymi złogami zlokalizowanymi centralnie, z przejrzystą rogówką zlokalizowaną na obwodzie i przejrzystą rogówką pomiędzy złogami (patrz rysunek 3A i 3B). W początkowym stadium choroba przebiega bezobjawowo, ale z czasem zmiany mogą się powiększać i prowadzić do pogorszenia widzenia. Nawracające nadżerki rogówki mogą wystąpić w GCD, ale z mniejszą częstością niż w LCD (1, 5). Pacjenci mogą również doświadczać olśnienia i światłowstrętu (2). Leczenie na wczesnym etapie procesu chorobowego polega często wyłącznie na obserwacji. Jednak w miarę postępu choroby może być konieczne zastosowanie PTK i przeszczepu rogówki w celu poprawy widzenia i erozji objawów. Podobnie jak w przypadku LCD, choroba może nawracać w przypadku przeszczepów rogówki.
Histopatologicznie zmiany mają postać eozynofilowych złogów, często opisywanych jako „rock candy like” w zrębie przednim, wykonanych z materiału podobnego do hialiny. Z czasem złogi te przechodzą do głębszego zrębu rogówki. Materiał hialinowy barwi się na jasnoczerwono przy użyciu barwnika trichromowego Massona (patrz rysunek 3C i 3D).
| A:Zdjęcie w lampie szczelinowej ziarnistej dystrofii rogówki, typ I | B: Zwrócić uwagę na „podobne do okruchów” złogi zrębu z wyraźnym interweniującym zrębem. |
 |
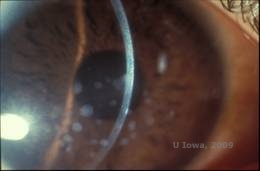 |
| C: Barwienie H& E rogówki ukazujące eozynofilowe złogi hialinowe w zrębie „przypominające cukierki skalne” | D: Hyaline material stains bright red with Masson-Trichrome |
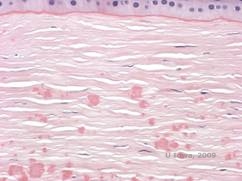 |
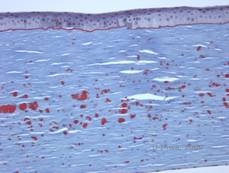 |
GRANULAR CORNEAL DYSTROPHY, TYPE II
Granular corneal dystrophy, type II (GCD2), znana wcześniej jako Avellino lub kombinowana ziarnisto-warstwowa dystrofia rogówki, jest chorobą dziedziczoną w sposób autosomalny dominujący, związaną z mutacją w genie TGFβI, która prowadzi do odkładania się zarówno hialiny, jak i amyloidu w zrębie rogówki. Zazwyczaj pacjenci pojawiają się w drugiej dekadzie życia z małymi szarobiałymi kropkami w powierzchownym zrębie rogówki. Zmętnienia mogą mieć kształt ciernisty, pierścieniowy lub gwiaździsty. W retroiluminacji są one częściowo przezierne. W późniejszym okresie procesu chorobowego mogą pojawić się w nich również linie siatkowate (patrz rysunek 4A i 4B). Linie te nie krzyżują się ze sobą i wydają się bielsze i mniej refrakcyjne niż linie siatkowate. Objawami GCD2 są ból z nadżerkami nabłonka i zaburzenia widzenia (2).
Histopatologicznie, rogówka będzie miała złogi barwiące się na czerwono za pomocą trichromu Massona, wskazujące na obecność hialiny (patrz rysunek 4C). Dodatkowo, barwienie czerwienią Kongo wykaże jabłkowo-zieloną dwójłomność przy polaryzacji krzyżowej, wskazując na obecność amyloidu (patrz Rysunek 4D). Uważano, że choroba wywodzi się od rodziny z Avellino we Włoszech. Jednakże, GCD typu II jest obecnie opisywana u pacjentów z wielu innych krajów (2,5), z największą częstością występowania we wschodniej Azji.
| A: Dystrofia Avellino przedstawiająca złogi podobne do siatki i podobne do ziarnistości w zrębie rogówki | B. Barwienie Massona Trichroma wykazujące złogi hialinowe w przednim odcinku zrębu |
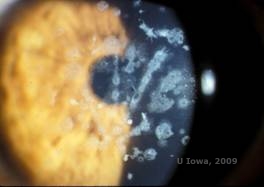 |
 |
| C: Barwienie czerwienią Kongo wykazujące różowe, amorficzne złogi amyloidowe w tym samym preparacie rogówki. | D: Polaryzacja krzyżowa ujawnia zieloną birefleksję wskazującą na amyloid. |
 |
 |
STROMALNA DYSTROFIA ROGÓWKI
MACULARNA DYSTROFIA ROGÓWKI
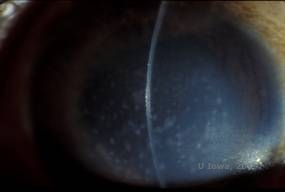
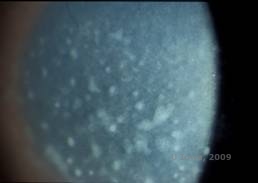
Macularna dystrofia rogówki (ang. (MCD) to choroba autosomalna recesywna spowodowana mutacją w genie sulfotransferazy węglowodanowej 6 (CHST6) na chromosomie 16, która prowadzi do defektu w syntezie siarczanu keratanu, głównego glikozaminoglikanu rogówki. Występuje rzadziej niż LCD lub GCD, ale ma tendencję do poważniejszego wpływu na wzrok. Chociaż MCD występuje na całym świecie rzadziej niż LCD czy GCD, jest najczęstszą dystrofią zrębu rogówki w takich miejscach jak Islandia czy Arabia Saudyjska (2,6). Szaro-białe, plamiste przednie zmiany stromalne podobne do GCD1 pojawiają się w rogówce w pierwszej dekadzie życia. Jednak w przeciwieństwie do GCD1, pomiędzy złogami występuje zamglenie zrębu, a cała rogówka od limbusu do limbusu jest często zajęta (patrz rysunek 5A i 5B). Rogówka jest cienka, a w miarę postępu choroby błona Descemeta staje się szara i pojawiają się w niej guttae. Może dojść do erozji nabłonka, ale w mniejszym stopniu w MCD niż w LCD. U pacjentów w drugiej lub trzeciej dekadzie życia dochodzi do poważnej utraty wzroku z powodu rozproszonego zamglenia rogówki. PTK może być przeprowadzony w niektórych wczesnych przypadkach MCD. Jednakże, stan ten generalnie nie jest tak podatny na PTK jak dystrofia siatkowata lub ziarnista i często wymaga przeszczepu rogówki w celu leczenia (7). Nawroty w przeszczepach występują rzadziej w MCD niż w przypadku dystrofii ziarnistej lub łaciatej (1,2,5,6,8).
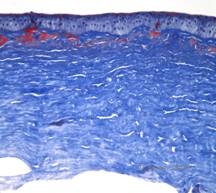
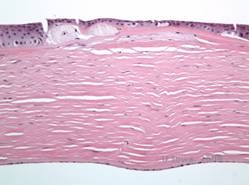
Złogi zrębu w MCD składają się z mukopolisacharydów, które gromadzą się w retikulum endoplazmatycznym keratocytów zrębu rogówki, pozakomórkowo pomiędzy blaszkami zrębu oraz w obrębie nabłonka, błony Descemeta i śródbłonka. Złogi te barwią się na niebiesko błękitem Alciana (patrz rysunek 5C i 5D) (1). Występują przerwy w warstwie Bowmana i guttae z pogrubieniem błony Descemeta (2).
Trzy podtypy MCD zostały opisane na podstawie obecności lub braku immunoreaktywnego siarczanu keratanu w różnych tkankach. Typ I nie wykazuje immunoreaktywnego siarczanu keratanu w zrębie rogówki, keratocytach, surowicy lub chrząstce i jest najczęstszym wariantem MCD na świecie. Typ IA nie ma siarczanu keratanu w zrębie, surowicy i chrząstce, ale ma wykrywalne poziomy wewnątrz keratocytów. Typ II ma siarczan keratanu obecny w znacznie zmniejszonej ilości w zrębie, keratocytach, surowicy i chrząstce (6).
| A: Zdjęcie w lampie szczelinowej makularnej dystrofii rogówki. | B: Zwróć uwagę na zamglenie pomiędzy złogami zrębu rogówki |
 |
 |
| C: H&E rogówki z dystrofią plamki. Należy zwrócić uwagę na przednie złogi stromalne i przerwanie warstwy Bowmana | D: Złogi mukopolisacharydów w obrębie keratocytów podkreślone barwnikiem Alcian Blue |
 |
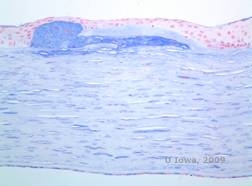 |
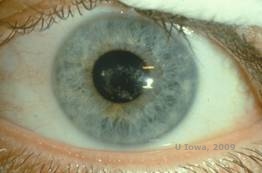
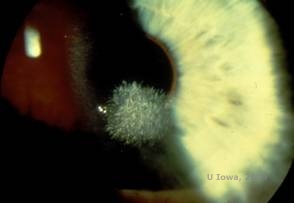
DYSTROPIA ROGÓWKI SCHNYDERA (SCD)
Dystrofia rogówki Schnydera (SCD), znana wcześniej jako krystaliczna dystrofia rogówki Schnydera, jest dziedziczoną autosomalnie dominująco, obustronną dystrofią zrębu rogówki związaną z mutacją genetyczną w genie UbiA zawierającym domenę prenylotransferazy 1(UBIAD1) na chromosomie 1. Wynikający z tego defekt metaboliczny keratocytów rogówki prowadzi do odkładania się krystalicznego cholesterolu w zrębie. Obecność kryształów nie jest jednak bezwzględnie konieczna do rozpoznania SCD. W rzeczywistości, tylko 54% pacjentów z SCD ma kryształy w rogówce. Zazwyczaj pacjenci pojawiają się w drugiej lub trzeciej dekadzie życia z pierścieniowatym centralnym zmętnieniem rogówki z lub bez przecinkowatych podnabłonkowych kryształów (patrz rysunek 6A i 6B). Następnie, między 23 a 38 rokiem życia pojawia się arcus lipoides. Po 38 roku życia postępujące zmętnienie rogówki prowadzi do panstromalnego zamglenia sięgającego do środkowego obwodu. U większości pacjentów po 50 roku życia dochodzi do fotopowej utraty wzroku, olśnienia i osłabienia czucia rogówki, dlatego mogą oni wymagać leczenia operacyjnego, w tym przeszczepu rogówki lub PTK. Może dojść do nawrotu choroby w przeszczepie. Choroba jest związana z hipercholesterolemią, hiperlipidemią i genu valgum u niektórych pacjentów (2,5,9,10).
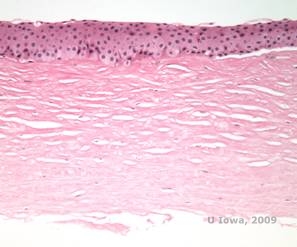
Histopatologicznie, dwubarwne kryształy cholesterolu składające się z fosfolipidów i cholesterolu odkładają się w komórkach nabłonka podstawnego, keratocytach, warstwie Bowmana i pomiędzy blaszkami zrębu. Lipidy rozpuszczają się w normalnej obróbce histologicznej, dlatego należy uzyskać mrożone wycinki rogówki, aby wykazać obecność lipidów za pomocą barwników Oil-Red-O lub czerni sudanowej.
| A: Zdjęcie w lampie szczelinowej dystrofii rogówki Schnydera. | B. Centralnie położone złogi krystaliczne |
 |
 |
| C: H&E rogówki z SCCD | D. Barwnik Oil Red O uwidacznia kryształy cholesterolu, które wyglądają na czerwone. |
 |
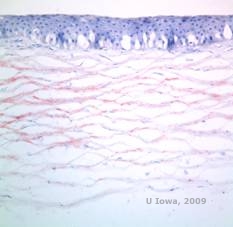 |
Tabela 4 dostarcza wspólnego mnemotechnika do zapamiętania niektórych dystrofii rogówki wpływających na zrąb, skład ich złogów, a metoda barwienia tych złogów jest wymieniona.
Tabela 4: Mnemonik do zapamiętania dystrofii zrębu rogówki
- Dystrofia Marilyn-Macular
- Monroe-Mucopolysaccharide
- Always-Alcian Blue stain
- Gets-Granular Dystrofia
- Her-Hyaline
- Man in-Masson Trichrome stain
- Los-Lattice Dystrophy
- Angeles-Amyloid
- California-Congo Red
OVERVIEW: CORNEAL STROMAL DYSTROPHIES
EPIDEMIOLOGIA
|
ZNACZENIA
|
SYMPTOMY
|
Postępowanie
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Conjunctival and Corneal Pathology. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3rd edition. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D classification of corneal dystrophies–edition 2. Cornea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. Clinical and genetic aspects of the TGFBI-associated corneal dystrophies. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nature genet. 1994; 6: 47-51.
- Bron AJ. Genetics of the Corneal Dystrophies: What we have learned in the past twenty five years. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating Keratoplasty for Macular Corneal Dystrophy. Ophthalmology 2005;112: 220-224.
- Badr IA, Wagoner MD. Phototherapeutic Keratectomy for Macular Corneal Dystrophy. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetics of anterior and stromal corneal dystrophies. Seminars in Ophthalmology, 2008; 23: 1,9-17.
- Weiss JS. Schnyder corneal dystrophy. Curr Opin Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. The gene for Schnyder’s crystalline corneal dystrophy maps to human chromosome 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Sugerowany format cytowania: Van C, Syed NA. Epithelial-Stromal and Stromal Corneal Dystrophies: A Clinicopathologic Review. Revision of ; EyeRounds.org. August 20, 2015. Dostępne od: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm