- Contexte
- Physiologie normale et physiopathologie du potassium
- Potentiel d’action d’un cardiomyocyte non pacemaker
- Potentiel d’action d’une cellule pacemaker cardiaque
- Conduction du courant
- Période réfractaire
- Hyperkaliémie, classification et causes
- Classification
- Causes
- Effets de l’hyperkaliémie
- Effets métaboliques
- Pompe sodium-potassium
- Conclusion
Contexte
Le potassium est un cation mou, blanc argenté, très réactif, appartenant à la famille des métaux alcalins du tableau périodique. C’est le cation le plus abondant dans le corps humain dans son ensemble, et l’ion le plus répandu dans ses compartiments intracellulaires.
En moyenne, un régime alimentaire occidental contient de 80 à 100 mEq de potassium par jour, et dans des conditions physiologiques normales, 90% de celui-ci est absorbé passivement, ne laissant que 9,0 mmol pour l’excrétion fécale. Les 3500-4000 mmol conservés dans le corps sont disproportionnés par rapport aux niveaux de potassium plasmatique diurnes qui sont normalement maintenus dans la gamme de 3,5-5,3 mmol/L par des mécanismes d’homéostasie serrés, les niveaux les plus bas étant la nuit et dans les premières heures du matin et le niveau maximal le plus élevé dans les heures de l’après-midi.
Une fois absorbé dans la circulation sanguine, le rôle du rein devient de faire correspondre l’apport en potassium à la sortie de potassium ; ce qui nécessite plusieurs heures, pendant lesquelles la » balance interne du potassium » sous l’influence de l’insuline et des catécholamines maintient une homéostasie temporaire en déplaçant le potassium entre les espaces intracellulaire et extracellulaire. La stimulation des récepteurs alpha empêche l’entrée du potassium dans les cellules, et la stimulation des récepteurs bêta la favorise en activant la pompe sodium-potassium ATPase.
La pompe sodium-potassium ATPase est l’enzyme gardienne de la porte située dans le sarcolemme. Elle permet de sauvegarder 98 % du potassium (environ 144,0 mmol) retenu à l’intérieur de la cellule. Cela assure la préservation de la différence de potentiel vitale à travers les membranes cellulaires, nécessaire au bon fonctionnement des cellules, en particulier les cellules excitables comme les cellules nerveuses et les cellules du muscle cardiaque.
Physiologie normale et physiopathologie du potassium
Après son absorption rapide, le potassium contribue à orchestrer ses propres niveaux corporels par la libération d’insuline et d’aldostérone. D’autres stimuli corporels inhérents se sont également avérés contrôler les niveaux corporels de potassium, notamment les récepteurs bêta-2 adrénergiques, le PH sanguin alcalin et l’anabolisme cellulaire.
Libération d’insuline et d’aldostérone : Le potassium ingéré passe rapidement dans la circulation. En atteignant la circulation portale, il stimule le pancréas à libérer de l’insuline. Simultanément, le potassium circulant atteignant les cellules juxtaglomérulaires entraîne la libération de rénine. La rénine, en atteignant le foie, est convertie en angiotensine I. L’angiotensine I se déplace vers les poumons où elle est convertie en angiotensine II. L’angiotensine II termine ensuite son voyage de retour vers les reins par le sang circulant pour stimuler la zone glomérulaire à sécréter l’aldostérone.
Equilibre interne du potassium : L’insuline libérée en post-prandial agit principalement sur les muscles squelettiques en activant deux voies, la voie AKT-dépendante responsable de l’insertion du transporteur de glucose GLUT4 et la voie APK activant l’ATPase sodium potassium cellulaire pour déplacer le potassium dans l’espace intracellulaire. Contrairement à la voie AKT-dépendante, la voie APK n’est altérée ni par le syndrome métabolique ni par la maladie rénale chronique (figure 1).
Excrétion : Le potassium filtré par les glomérules rénaux est réabsorbé passivement dans le tubule proximal et l’anse de Henle, proportionnellement à la quantité de sodium et d’eau apportée. Normalement, seulement environ 10% de la charge filtrée atteint le néphron distal.
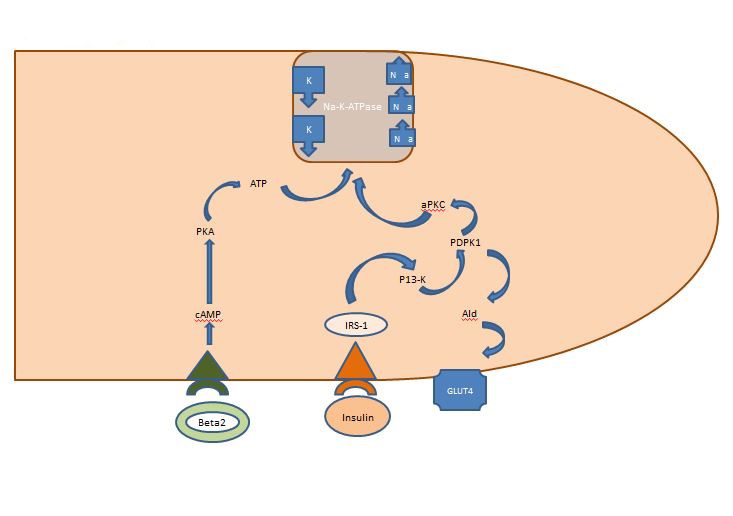
Figure 1. Action de l’insuline sur une cellule de muscle squelettique. L’insuline libérée post-prandiale active deux voies dans les muscles squelettiques, la voie AKT-dépendante responsable de l’insertion du transporteur de glucose GLUT4 et la voie APK activant l’ATPase sodium potassium cellulaire pour déplacer le potassium dans l’espace intracellulaire.
Au début du tubule contourné distal, la sécrétion de l’excès de potassium commence et augmente progressivement en avançant vers le néphron distal et dans le canal collecteur. Ceci est médié par la régulation à la hausse de l’hydrogène potassium ATPase sur les cellules alpha-intercalées .
La présence de niveaux de potassium plus élevés dans les cellules péritubulaires des reins active le système SRAA pour libérer l’aldostérone, qui active l’ATPase sodium potassium dans la membrane basolatérale, ce qui entraîne une diminution du sodium intracellulaire qui conduit à l’augmentation du transport électrogène de l’absorption du potassium en hyperpolarisant le voltage de la membrane et en permettant son excrétion dans l’urine .
En cas d’hyperkaliémie, le quota de potassium excrété par le côlon peut augmenter jusqu’à 30%, par ex, en cas d’insuffisance rénale, où le potassium est alors activement capté par la pompe sodium potassium ATPase activée dans la membrane basolatérale des entérocytes coliques, pour être excrété de l’autre côté, dans la lumière colique par les grands canaux potassiques apicaux calcium-dépendants des cellules.
Il est donc discernable de ce qui précède que le mécanisme de l’homéostasie du niveau plasmatique de potassium est ordonné principalement par l’interaction de trois transactions simultanées – l’apport de potassium, les déplacements intra/extracellulaires de potassium et l’excrétion urinaire de potassium, qui reposent tous finalement sur la pompe sodium-potassium.
Pour comprendre le mécanisme du danger imminent de l’hyperkaliémie et sa gestion, il faut comprendre la physiologie du potentiel d’action et les entrailles de l’enzyme sodium potassium ATPase.
Electrophysiologie du potentiel d’action, c’est-à-dire , mouvement ionique à travers les membranes cellulaires, est déterminée par la différence de deux potentiels, un « potentiel chimique » dans lequel les ions se déplacent le long de leur gradient de concentration et un « potentiel électrique » dans lequel les ions et les molécules repoussent les charges semblables, ce qui donne le potentiel transmembranaire (PTM), qui est dit +ve lorsque le mouvement net des ions +ve se fait vers l’extérieur de la cellule et vice versa.
Potentiel d’action d’un cardiomyocyte non pacemaker
Le potentiel d’action comporte cinq phases, qui commencent et se terminent à la phase 4. Les pompes impliquées dans ce processus comprennent l’échangeur sodium-calcium du sarcolemme, l’ATPase calcique et, finalement, l’ATPase sodium-potassium.
- Phase 4. La phase de repos : elle présente un potentiel de repos de -90 mV en raison du mouvement constant du potassium vers l’extérieur via les canaux redresseurs vers l’intérieur. Pendant cette phase, les canaux sodiques et calciques sont fermés.
- Phase 0. La phase de dépolarisation : l’allumage d’une cellule pacemaker ou sa conduction par une cellule voisine déclenche l’élévation du TMP au-dessus de -90 mV. À ce stade, les « canaux sodiques rapides » commencent à s’ouvrir un par un, permettant au sodium de pénétrer dans la cellule, ce qui fait monter la TMP et, une fois que suffisamment de canaux sodiques rapides se sont ouverts pour atteindre -70 mV, un courant sodique entrant auto-entretenu est mis en marche, dépolariser rapidement le TMP à 0 mV pour un intérim transitoire connu sous le nom de « overshoot », à ce moment-là, les canaux sodiques rapides dépendant du temps se ferment et les canaux calciques « à ouverture longue » s’ouvrent pour élever le TMP à -40 mV et permettre un petit afflux régulier de calcium le long de son gradient de concentration.
- Phase 1. La phase de repolarisation précoce : elle commence avec le TMP légèrement +ve et la brève ouverture de certains canaux potassiques entraînant son écoulement vers l’extérieur de la cellule, ramenant le TMP à environ 0 mV.
- Phase 2. La phase de plateau : ici les deux contre-courants sont électriquement équilibrés et aboutissent au maintien du TMP équilibré juste en dessous de 0 mV. « Les canaux calciques à ouverture longue » sont toujours ouverts, ce qui entraîne un flux constant de calcium dans la cellule. Le canal potassique à redressement différé permet le passage du potassium vers l’extérieur de la cellule en descendant son gradient de concentration.
- Phase 3. La phase de repolarisation : au cours de cette phase, les canaux calciques sont progressivement inactivés et le flux persistant de potassium vers l’extérieur de la cellule dépasse ainsi le flux calcique entrant, renvoyant le potassium vers l’espace intracellulaire et le sodium et le calcium vers l’extérieur de la cellule.
Potentiel d’action d’une cellule pacemaker cardiaque
Les cellules pacemakers cardiaques ont une automaticité innée, permettant leur dépolarisation en cycles rythmiques. Le nœud sinusal (SAN) a le rythme de dépolarisation auto-initié le plus élevé, à une fréquence de 60-90/min, suivi du nœud auriculo-ventriculaire (AVN) à une fréquence de 40-60/min, puis des fibres de Purkinje et du muscle ventriculaire à 20-40/min.
Les potentiels membranaires des cellules pacemaker sont instables et leurs potentiels d’action n’ont pas de phases bien définies. Elles ont moins de canaux potassiques à rectification vers l’intérieur et leur TMP ne descend jamais en dessous de -60 mV, éliminant le rôle des canaux sodiques rapides qui nécessitent un TMP de -90 mV résultant en l’absence de la phase de dépolarisation rapide.
Au TMP >-60 mV, le courant « funny/pacemaker » est mis en action avec un flux spontané d’ions à travers les canaux sodiques lents, dépolarisant le TMP à <-50 mV puis revenant à -60 mV lorsque les canaux calciques se ferment.
Conduction du courant
Tous les cardiomyocytes sont couplés électriquement par la jonction gap, y compris la cellule pacemaker. Cela facilite la dépolarisation généralisée de toutes les cellules voisines, transformant le cœur en une unité fonctionnelle dans laquelle la cellule ayant la fréquence inhérente la plus élevée devient le « pacemaker ».
Période réfractaire
La période réfractaire plus longue pendant le long plateau de la phase 2, due aux canaux calciques lents, fournit le temps nécessaire à la vidange complète des ventricules avant la contraction suivante. Les périodes réfractaires peuvent être absolues (ARP), effectives (ERP) ou relatives (RRP). Dans une PRA, la cellule est absolument non excitable.
Une PRE dure de la PRA jusqu’au segment court de la phase 3. Un stimulus à ce stade pourrait dépolariser minimalement la cellule, mais le niveau de dépolarisation est plus faible que la propagation d’un potentiel d’action aux cellules voisines.
La PRR est provoquée par un stimulus supérieur à la normale, entraînant la dépolarisation de la cellule et la production d’un potentiel d’action.
Une « période supra-normale » est un état hyperexcitable pendant lequel un stimulus plus faible que la normale pourrait conduire à une arythmie, nécessitant la synchronisation pendant la cardioversion pour éviter la fibrillation ventriculaire (figure 2).
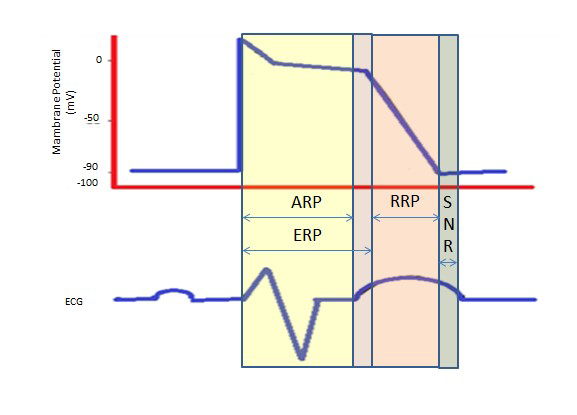
Figure 2. Périodes réfractaires. ARP : Période réfractaire absolue ; PRE : Période réfractaire effective ; PRR : Période réfractaire relative ; SNR : Période réfractaire supranormale
Hyperkaliémie, classification et causes
Classification
L’hyperkaliémie est classée comme légère lorsque les niveaux sont dans la gamme de 5,5-6,0 mmol/L, modérée de 6,1-6,9 mmol/L et sévère à des niveaux de 7.0 mmol/L ou plus, et à tout niveau auquel des modifications de l’ECG apparaissent.
Causes
L’hyperkaliémie survient lorsque les mécanismes compensatoires ne sont plus en mesure de faire face au déséquilibre, c’est pourquoi elle est généralement multifactorielle.
- Augmentation de l’apport en potassium par n’importe quelle voie, par ex, apport alimentaire oral, ou administration intraveineuse de liquides contenant du potassium comme la pénicilline G.
- Rétention par les reins : étant donné que l’excrétion du potassium dépend de l’aldostérone et de la délivrance d’une quantité distale suffisante de sodium et d’eau dans les néphrons, des conditions telles que l’insuffisance rénale, l’insuffisance surrénale (maladie d’Addison) , l’hypoaldostéronisme hyporéninémique de type IV, l’acidose tubulaire rénale, en particulier chez les patients atteints de néphropathie diabétique ainsi que toute condition qui favorise l’hypoperfusion comme dans la déplétion volumique et l’insuffisance cardiaque congestive, affecteront l’équilibre complexe du potassium dans le corps et prédisposeront à l’hyperkaliémie.
- Insuffisance surrénalienne : elle doit être exclue chez les patients hyperkaliémiques, notamment en présence d’une hyponatrémie et d’une faiblesse musculaire. Pour dépister une insuffisance surrénale primaire, un test standard de stimulation de la cosyntropine est effectué : 0,25 mg de cosyntropine synthétique est administré en bolus intraveineux, suivi d’une mesure du cortisol plasmatique 45 minutes à 1 heure plus tard. Des valeurs inférieures à 20 mcg/dL sont évocatrices d’une insuffisance surrénale.
- Médicaments qui retiennent le potassium : les médicaments délivrés sur ordonnance qui réduisent l’activité de l’ATPase sodium-potassium, tels que les bêtabloquants, et les médicaments qui réduisent la sécrétion d’aldostérone, tels que les inhibiteurs de l’ECA et de l’ARA, les anti-inflammatoires non stéroïdiens et les diurétiques d’épargne potassique, doivent faire l’objet d’un suivi étroit pour éviter une hyperkaliémie iatrogène, en particulier dans le groupe d’âge gériatrique avec leur déclin progressif de la fonction rénale dans le cadre du processus de vieillissement.
- Perturbations du déplacement transcellulaire du potassium : cela peut se produire dans des conditions d’acidose, d’hyperglycémie, d’hyperosmolalité, d’exercice intense, de dégradation des tissus, de paralysie périodique hyperkaliémique et avec les bêtabloquants. Pour chaque diminution de 0,1 unité du PH sanguin, le potassium sérique augmente d’environ 0,6 mmol/L (moins si l’acidose est causée par des acides organiques) .
- Le pseudo-hypoaldostéronisme est une maladie congénitale autosomique récessive dans laquelle les reins sont résistants aux actions de l’aldostérone.
- La pseudo-hyperkaliémie ne doit pas non plus être négligée : comme son nom l’indique, elle se manifeste par une élévation du potassium sérique en présence d’un potassium plasmatique normal. Elle peut être observée dans le cas de sang hémolysé, d’un garrot serré prolongé pendant une procédure de prélèvement sanguin, provoquant la libération extracellulaire de potassium, en cas de serrage répété du poing pendant une phlébotomie, d’une ponction veineuse traumatique, en cas de leucocytose et de thrombocytose, et dans certains syndromes génétiques peu courants tels que la pseudo-hyperkaliémie familiale et la sphérocytose héréditaire. Cependant, elle pourrait tout simplement résulter d’une simple erreur de laboratoire.
Effets de l’hyperkaliémie
L’hyperkaliémie légère est souvent asymptomatique, détectée accidentellement par des tests de laboratoire, en raison de ses symptômes vagues comme le malaise, la faiblesse musculaire et la paresthésie. Une hyperkaliémie sévère affectera la fonction neuromusculaire sous la forme d’une faiblesse des muscles squelettiques et d’une paralysie ; cependant, ce n’est pas une présentation fréquente car la toxicité cardiaque domine le tableau et constitue la présentation préliminaire. La toxicité cardiaque se présentera généralement sur l’ECG de la manière suivante, en escalade, bien que ce ne soit pas nécessairement le cas, selon l’étiologie :
- À des niveaux supérieurs à 5,5 mEq/L, l’augmentation de la conductance des canaux potassiques augmente le courant lkr, ce qui entraîne une repolarisation rapide sous la forme d’une onde T en pointe sur l’ECG de surface. Ces ondes T peuvent être différenciées de celles de l’infarctus du myocarde et de l’AVC par leur courte durée allant de 150 à 250 msec.
- À des taux de potassium supérieurs à 6,5 mEq/L, un état de dépolarisation soutenue sous-seuil se produit, entraînant un retard dans la dépolarisation auriculaire et ventriculaire. La diminution de la phase 0 du potentiel d’action conduit à un potentiel d’action plus long, produisant un retard dans la conduction intraventriculaire et auriculo-ventriculaire. Sur l’ECG de surface, cela se traduit par un aplatissement et une perte des ondes P et un élargissement des complexes QRS. Avec un retard croissant dans la conduction intraventriculaire, l’ECG de surface commence à montrer des signes de bloc de branche gauche et droit. Ce phénomène peut être différencié de la maladie du faisceau par le fait qu’en cas d’hyperkaliémie, le retard persiste tout au long du complexe QRS, et pas seulement pendant la partie initiale ou terminale, respectivement.
- À 10 mEq/L, la conduction sino-atriale ne se produit plus et le rythme jonctionnel accéléré prend le relais. Les arythmies ventriculaires se développent avec la fusion des complexes QRS élargis avec les ondes T pour finalement former le schéma sinusoïdal classique. Une fois que cela se produit, la FV et l’asystolie sont imminentes et l’arrêt cardiaque s’ensuit alors.
- Parfois, les changements peuvent être erratiques et imprévisibles et l’ECG sautera de la normale à l’asystole en raison de la variabilité des facteurs étiologiques et de leurs effets influents, par exemple, la vitesse de changement du potassium, la concentration de calcium, le pH et la concentration de sodium. Ainsi, l’hyperkaliémie doit être traitée d’urgence dès que le taux de potassium devient supérieur à 6,5 mmol/L, ou en présence de manifestations ECG d’hyperkaliémie quel que soit le taux de potassium. D’autres associations rapportées avec l’hyperkaliémie aiguë incluent : image de pseudo MI sur l’enregistrement ECG, avec un segment ST-T massif résultant de dérèglements de la repolarisation des myocytes, intervalles PR et QT courts, tachycardie sinusale, bradycardie sinusale, rythme idioventriculaire, bloc cardiaque du 1er et du 2ème degré .
Effets métaboliques
L’hyperkaliémie entraîne une acidose métabolique hyperchlorémique car l’hyperkaliémie favorise la captation intracellulaire de potassium en échange d’ions hydrogène. Cela crée une alcalose intracellulaire, supprimant la production rénale d’ammoniac dans les tubules proximaux, entraînant une diminution de l’excrétion urinaire d’ammonium et d’acide et une acidose tubulaire rénale de type IV .
Pompe sodium-potassium
L’ATPase sodium-potassium a été découverte en 1957 par Skou, qui a ensuite reçu une part du prix Nobel de chimie 1997 pour sa découverte.
Skou a été le premier à découvrir l’ATPase sodium-potassium dans le sarcolemme de la surface cellulaire des muscles cardiaques. Sa présence a ensuite été détectée dans tous les organismes eucaryotes unicellulaires et multicellulaires.
La pompe sodium potassium fonctionne en liant l’hydrolyse de l’ATP à l’exportation cellulaire de trois ions sodium en échange de deux ions potassium contre leurs gradients électrochimiques. C’est la cible moléculaire de la digitaline et de la digoxine, utilisées depuis le 18e siècle sous forme d’extraits de digitale.
L’action de la pompe sodium potassium est régulée par une phosphoprotéine, le phospholemman, dont la non-phosphorylation entraîne l’inhibition de la pompe et dont la phosphorylation entraîne une augmentation de l’activité de la pompe. Il possède trois sites de phosphorylation, deux sites de palmitoylation et un site de glutathionylation, ce qui explique la multitude de signaux capables de stimuler et d’inhiber la pompe.
La pompe sodium potassium est elle-même une enzyme composée de plusieurs sous-unités avec plusieurs isoformes. La présence des sous-unités alpha et bêta (principalement B1 dans le cœur) est essentielle pour sa fonction. Récemment, une troisième sous-unité protéique gamma a été identifiée dans les reins, mais à ce jour, sa fonction reste inconnue.
La sous-unité alpha est le noyau catalytique de l’enzyme de la pompe sodium potassium. Elle mesure environ 100 kDa et contient les sites de liaison pour le sodium, le potassium, l’ATP et les stéroïdes cardiotoniques comme l’ouabaïne. Seules les protéines alpha 1 et alpha 2 sont présentes de manière significative dans un myocyte cardiaque normal et sont fonctionnellement liées à l’échangeur sodium-calcium (NCX). On a signalé que l’alpha 3 remplace l’alpha 2 dans les modèles expérimentaux d’insuffisance cardiaque.
Les données d’expériences récentes favorisent l’implication des deux sous-unités alpha 1 alpha 2 de la pompe dans la régulation du couplage excitation-contraction (E-C). L’alpha 1, qui s’est avérée être plus uniformément distribuée à travers le sarcolemme, est censée jouer davantage un rôle de « ménage », contrôlant à la fois la contractilité et la masse de sodium intracellulaire, tandis que l’alpha 2 dont l’expression est concentrée dans les tubules T avec d’autres composants clés du couplage E-C est censée se concentrer principalement sur la contractilité .
Les facteurs connus qui peuvent contrôler la pompe sodium-potassium comprennent : ATP, sodium intracellulaire, barrières sub-sarcolemmales et espaces flous, potentiel membranaire, voies de signalisation intracellulaires (voies de signalisation adrénergiques, protéine kinase A & C, oxyde nitrique, phospholemmane), régulation directe par de petites molécules (lipides, stéroïdes cardiotoniques endogènes), autres protéines associées (cavéoles et cavéolines, et ankyrine).
Conclusion
L’hyperkaliémie est un défi clinique et peut se présenter chez jusqu’à 10% des patients hospitalisés . Son résultat final met en danger la vie du patient. Comme toutes les cellules du corps sont finalement affectées par la pompe sodium-potassium, et que les muscles cardiaques ischémiques sont connus pour extruder leur potassium extracellulairement, ce qui entraîne une réduction du seuil d’arythmie avec la possibilité d’arythmies ventriculaires qui aggravent l’hypopolarisation et abaissent encore plus le seuil, davantage d’études doivent être axées sur la manipulation de l’enzyme sodium-potassium, car son contrôle pourrait modifier favorablement les résultats des arrêts cardiaques et réécrire les directives actuelles de la RCP.