ABOVE : modifié de © ISTOCK.com, tera vector
Presque toujours, construire quelque chose est plus difficile que le démolir. De même, il est plus difficile d’introduire des gènes que de les supprimer. C’est une réalité que les chercheurs devront surmonter pour tirer le meilleur parti de l’édition de gènes. L’introduction de gènes permet aux scientifiques d’étudier les effets de variantes génétiques spécifiques, d’utiliser des gènes rapporteurs comme la protéine fluorescente verte pour suivre les produits géniques dans le temps et l’espace, de sonder la régulation du génome et, enfin, de réparer les gènes responsables de maladies. « C’est un moyen très efficace d’interroger chaque base d’un gène », déclare Greg Findlay, candidat au doctorat en médecine à l’université de Washington.
CRISPR-Cas9, une technologie d’édition de gènes connue pour sa convivialité, peut introduire ou supprimer des gènes. Le knocking out d’un gène implique l’insertion de CRISPR-Cas9 dans une cellule à l’aide d’un ARN guide qui cible l’outil sur le gène d’intérêt. Là, Cas9 coupe le gène, déchirant les deux brins d’ADN, et le mécanisme régulier de réparation de l’ADN de la cellule répare la coupure à l’aide d’un processus appelé jonction des extrémités non homologues (NHEJ). La NHEJ est très efficace mais imprécise. Le processus a tendance à introduire des erreurs sous forme de petites insertions ou délétions qui suffisent généralement à éliminer le gène.
Pour éliminer un gène, cependant, les coupures doivent être réparées très précisément, sans insertions ou délétions supplémentaires. Cela nécessite l’utilisation d’un second mécanisme de réparation de l’ADN appelé réparation dirigée par homologie (HDR), qui, dans les cellules de mammifères du moins, est moins efficace, de sorte que sa fréquence est éclipsée par celle du NHEJ. Le fait que certains loci de gènes et certains types de cellules soient intrinsèquement moins hospitaliers à l’édition CRISPR-Cas9 complique encore le processus.
Au cours des dernières années, les chercheurs ont développé de nombreuses nouvelles stratégies pour augmenter l’efficacité du knocking dans les gènes, petits et grands, à l’aide de CRISPR-Cas9, et en cours de route, ils ont proposé et testé de nouvelles applications pour ce type d’édition de gènes. Ici, The Scientist explore quelques-unes des approches les plus prometteuses.
Select It
Chercheur : Jon Chesnut, directeur principal de la biologie synthétique R&D, Thermo Fisher Scientific
Projet : En développant un kit de marquage de gènes appelé Truetag que Thermo Fisher mettra sur le marché plus tard cette année, Chesnut a utilisé des marqueurs sélectionnables pour améliorer
l’efficacité. Un marqueur sélectionnable – dans ce cas, un gène de résistance aux antibiotiques – est collé à une étiquette de protéine fluorescente et introduit dans des cellules de mammifères. Ces cellules sont ensuite cultivées avec l’antibiotique associé. Le gène de résistance confère un avantage sélectif aux cellules qui le portent ; elles sont les seules à pouvoir se développer, et donc celles qui se développent contiennent l’étiquette du gène en question. Même si l’efficacité de l’insertion du gène est faible, les chercheurs peuvent utiliser la sélection antibiotique pendant une semaine ou plus pour se retrouver avec un pourcentage élevé de cellules avec des insertions réussies.
En utilisant l’antibiotique puromycine ou blasticidine avec le kit, l’équipe de Chesnut a réussi à augmenter le taux d’insertion du gène de 10-30% à 90% ou plus dans certaines populations de cellules. Quelques gènes particulièrement difficiles sont passés d’un taux d’insertion de moins de 1 % à plus de 90 %. Il est important de tester plusieurs doses d’antibiotiques sur la lignée cellulaire que vous prévoyez d’utiliser pour trouver la bonne dose, dit Chesnut : vous voulez tuer les cellules sans insertions mais pas les cellules avec des insertions réussies.
Essayez-le : Les marqueurs sélectifs fonctionnent mieux lorsque le gène d’intérêt est fortement exprimé, dit Chesnut. « S’il ne l’est pas, vous pouvez quand même obtenir une sélection, mais vous risquez de ne pas obtenir une expression suffisante de votre étiquette de protéine fluorescente pour pouvoir la détecter. » En outre, les limites générales de CRISPR-Cas9 s’appliquent. « Il y a des régions du génome qui ne se coupent pas très bien avec CRISPR, et nous ne savons toujours pas pourquoi », ajoute-t-il. Et certains types de cellules n’acceptent pas facilement l’ADN, l’ARN ou les complexes ARN-protéine étrangers – les trois méthodes de livraison de CRISPR-Cas9.
Pour avoir plus de chance d’insérer des marqueurs sélectionnables, assurez-vous qu’il y a une séquence dite PAM, une courte étiquette dans l’ADN cible que CRISPR-Cas9 doit reconnaître avant de couper, dans un rayon de 10 paires de bases du site d’insertion du gène souhaité, dit Chesnut. Plus loin du site de coupe, l’efficacité de l’insertion peut être trop faible pour être fonctionnelle. Sans site PAM, vous pouvez essayer les TALEN ou les nucléases à doigt de zinc, bien que ces anciennes techniques d’édition de gènes soient plus délicates que CRISPR.
Inhibition programmée
Chercheur : Jacob Corn, biologiste du génome, École polytechnique fédérale de Zurich
Projet : Les chercheurs ne comprennent pas pourquoi la voie NHEJ surpasse largement la voie HDR dans les cellules de mammifères. « La levure fait de l’HDR comme une folle », dit Corn. Dans le but de relancer ce processus de réparation de l’ADN dans les cellules humaines et d’améliorer le contrôle de l’inactivation des gènes, son équipe et lui tentent de déterminer avec précision comment la HDR est régulée. Ils ont analysé des cellules humaines pour trouver des gènes dont l’élimination entraîne une augmentation de la réparation de l’ADN dans la cellule, puis ils ont recherché des petites molécules inhibitrices de ces gènes. L’un des gènes qui est apparu codait pour CDC7, une kinase qui régule la transition du cycle cellulaire vers la phase S ; son inhibiteur, XL413, a multiplié par deux ou trois l’efficacité de l’inactivation du gène (BioRXiv, DOI : 10.1101/500462, 2018). Cela s’explique par le fait que la HDR ne se produit que dans certaines parties du cycle cellulaire, notamment la phase S, explique Corn. Si vous ajoutez l’inhibiteur XL413 en même temps que vous utilisez CRISPR-Cas9 pour modifier votre gène cible, les cellules s’accumulent dans la phase immédiatement antérieure à la phase S. Ensuite, vous retirez XL413, et toutes les cellules passent en phase S et augmentent l’efficacité du knock-in.

Corn a utilisé cette technique dans de nombreuses lignées de cellules humaines immortalisées et dans des cellules T humaines. Elle peut frapper de courtes portions d’ADN, comme les SNP, ainsi que de grands gènes. Il n’y a aucune raison pour que cela ne fonctionne pas chez les souris, dit-il, bien qu’il ne l’ait pas testé.
Essayez-le : « Le timing est absolument clé », dit Corn. Le Cas9 doit couper l’ADN en même temps que le XL413 est ajouté. Si vous inhibez d’abord et libérez ensuite pendant l’édition avec CRISPR-Cas9, l’efficacité de la recombinaison homologue est divisée par trois au lieu d’augmenter, car les cellules sont libérées dans la mauvaise phase du cycle cellulaire.
Et comme pour tout effort d’HDR, Corn dit qu’il faut toujours exécuter un contrôle sans nucléase pour s’assurer que vous n’amplifiez pas accidentellement l’ADN contaminant qui flotte dans votre laboratoire. Après avoir introduit le knock-in, « séquencez, séquencez, séquencez, séquencez », dit-il. L’utilisation d’un système rapporteur tel qu’une étiquette de protéine fluorescente pour démontrer l’insertion réussie d’un gène peut se retourner contre vous. Le séquençage permet de vérifier que les insertions ont été faites au bon endroit.
Playing the Long Game
Chercheur : Channabasavaiah Gurumurthy, directeur du centre d’ingénierie du génome de la souris, Université du Nebraska Medical Center
Projet : Il y a quelques années, en réfléchissant à la difficulté d’assommer des gènes tout en essayant de le faire dans des zygotes de souris, Gurumurthy et ses collègues ont eu une révélation.
Les chercheurs réussissaient à insérer de l’ADN court et monocaténaire, alors pourquoi ne pas essayer de faire un knock-in en insérant de l’ADN long et monocaténaire ? En effet, l’approche, que Gurumurthy appelle Easi-CRISPR (additions efficaces avec des insertions d’ADN ss -CRISPR), augmente l’efficacité de 2,5 fois, et l’utilisation d’ADN simple brin réduit de 100 fois le taux d’insertions hors cible en culture cellulaire (Nat Protoc 13:195-215, 2018 ; Nature 559:405-09, 2018). » C’est assez énorme « , dit-il. Dans le laboratoire de Gurumurthy, Easi-CRISPR a généré une lignée de souris knock-in pour 9 gènes sur 10 qu’ils ont essayés. Un collaborateur l’a également utilisé dans des cellules T humaines pour créer des cellules CAR-T, des cellules immunitaires spécifiques aux patients pour combattre le cancer.
Essayez-le : Easi-CRISPR est loin d’être infaillible, prévient Gurumurthy. Parfois, la technique n’insère qu’une partie du gène. Il ajoute qu’elle peut aussi brouiller les bras d’homologie, c’est-à-dire les courtes séquences situées de part et d’autre du gène qui le relient à sa cible correcte dans le génome. Et certains loci sont inexplicablement plus difficiles à insérer que d’autres.
Peu de fournisseurs commerciaux conçoivent et synthétisent de longs ADN monocaténaires sur mesure. Vous pouvez fabriquer le vôtre, mais la stabilité de l’ADN simple brin varie ; les séquences moins stables auront des rendements plus faibles, de sorte que vous devrez peut-être en synthétiser davantage, dit Gurumurthy.
Les chercheurs qui ne peuvent pas insérer CRISPR dans des embryons de souris unicellulaires peuvent payer une installation centrale pour fabriquer les souris avec leur séquence d’ADN, dit Gurumurthy. Les installations de base comme la sienne facturent de 5 000 à 15 000 dollars pour générer une ou deux paires de reproduction ; les installations commerciales facturent de 20 000 à 50 000 dollars, dit-il.
Knock-in By Numbers
Chercheur : Greg Findlay, candidat MD/PhD dans le laboratoire de Jay Shendure, Université de Washington
Projet : Findlay et ses collègues avaient pour objectif d’améliorer la façon dont les cliniciens interprètent les mutations du gène BRCA1 du cancer du sein et de l’ovaire. Ce gène a des milliers de variantes, mais les chercheurs ne savent pas comment la plupart d’entre elles affectent sa fonction. Pour étudier l’impact de ces variants, ils ont utilisé une technique de knock-in qu’ils ont développée, appelée édition du génome par saturation (Nature, 562:217-22, 2018).
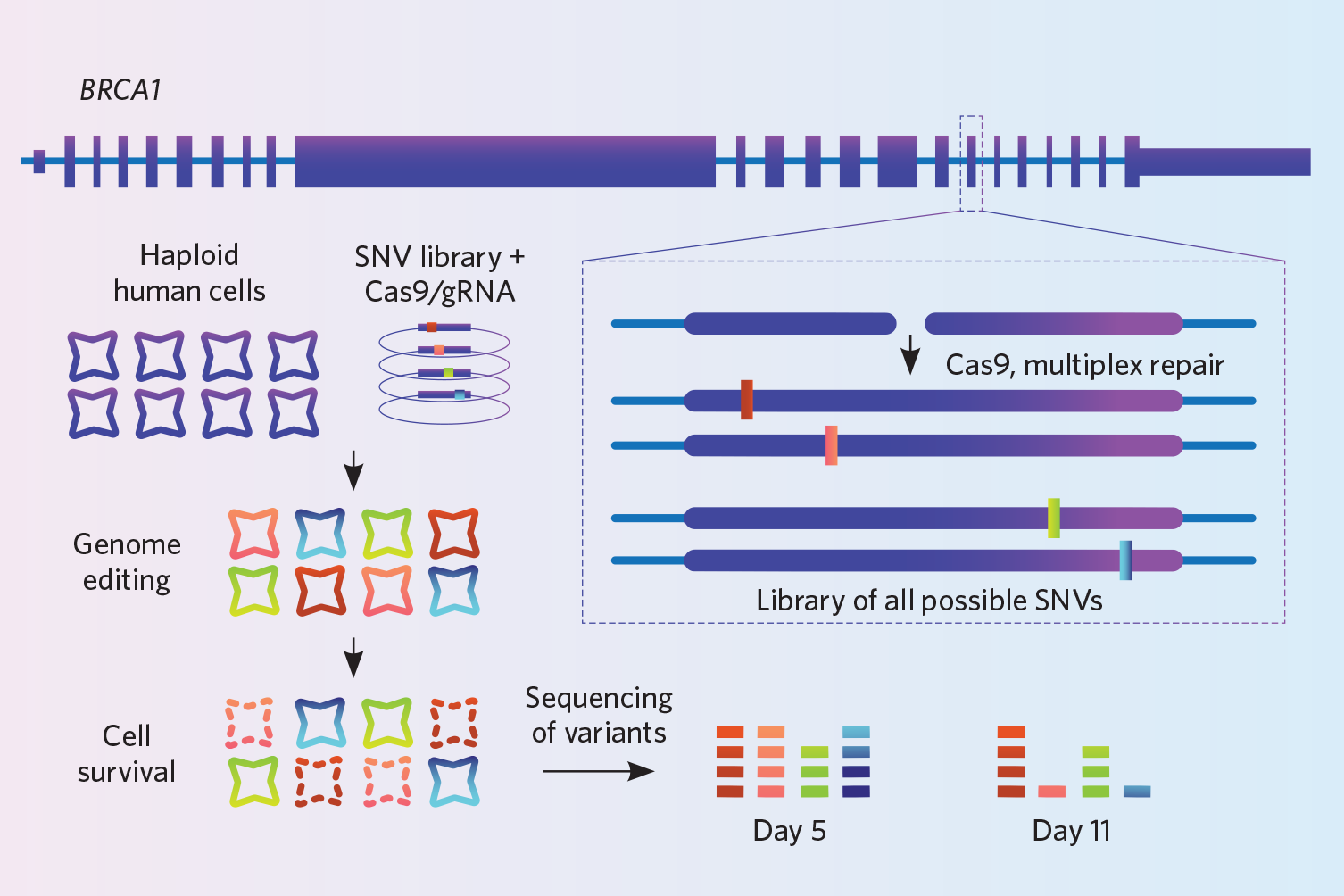
Dans une lignée de cellules humaines haploïdes immortalisées, ils ont utilisé CRISPR-Cas9 pour assommer 4 000 minuscules variants dans des millions de cellules à la fois in vitro. Le génome est coupé au même endroit dans chaque cellule, mais le génome de chaque cellule reçoit une variante différente. Pour favoriser l’HDR, les chercheurs ont également éliminé le gène de la ligase4, désactivant ainsi la voie de réparation NHEJ – une étape qui a permis de tripler l’efficacité, explique Findlay. Enfin, comme les knock-ins de toutes les cellules sont différents, ils ont séquencé les cellules en profondeur, couvrant la même région génomique des millions de fois, pour s’assurer qu’ils ont effectivement introduit les 4 000 variantes qu’ils voulaient étudier. Ils ont séquencé à deux moments, et en ont déduit que les knock-ins qui n’apparaissaient pas dans le séquençage au deuxième moment étaient ceux qui interféraient avec la fonction du gène, car les cellules qui les portaient devaient être mortes.
Essayez : L’équipe de Findlay a fait fabriquer pour elle les oligos d’ADN des 4 000 variantes sur un microréseau. Vous pouvez acheter des matrices de 6 000 à 250 000 oligos, alors envisagez d’en avoir plus pour votre argent en combinant plusieurs expériences sur la même matrice, dit Findlay. Leur laboratoire paie environ 5 000 $ pour 100 000 oligos.
Cette stratégie a ses limites : elle n’a été utilisée jusqu’à présent que pour éliminer des variantes d’un seul nucléotide, et toutes les modifications doivent être effectuées dans le même gène. La méthode fonctionne mieux lorsqu’il s’agit de modifier une région assez étroite de l’ADN, d’environ 110 à 120 paires de bases, car des oligos d’ADN plus longs comporteraient trop d’erreurs, explique Findlay. Il est également important de séquencer très profondément pour s’assurer que vous tenez compte du nombre total de variantes que vous aviez l’intention d’introduire.