- Une revue clinicopathologique
- INTRODUCTION
- Dystrophies épithéliales-stromales cornéennes
- DYSTROPHIE CORnéENNE DE REIS-BUCKLERS
- DYSTROPHIE CORNÉENNE EN RÉSEAU
- DYSTROPHIE CORNÉALE GRANULAIRE, TYPE I
- DYSTROPHIE CORNÉENNE GRANULAIRE, TYPE II
- DYSTROPHIES CORNÉALES STROMALES
- DYSTROPHIE CORNÉALE MACULAIRE
- DYSTROPHIE CORnéENNE DE SCHNYDER (DSC)
- Tableau 4 : Mnémonique pour se souvenir des dystrophies du stroma cornéen
- VISUALISATION : DYSTROPHIES CORNEALES STROMALES
- EPIDEMIOLOGIE
- SIGNES
- SYMPTÔMES
- TRAITEMENT
Une revue clinicopathologique
Emily S. Birkholz, MD, Nasreen A. Syed, MD, et Michael D. Wagoner, MD, PhD
17 août 2009
Révision majeure : Chaunhi Van, MD et Nasreen Syed, MD
Août 20, 2015
INTRODUCTION
Les dystrophies épithéliales-stromales et stromales cornéennes sont un groupe de troubles héréditaires de la cornée qui sont causés par l’accumulation progressive de dépôts dans les couches de la cornée. Ces dépôts ne sont pas causés par une inflammation, une infection ou un traumatisme, mais par des mutations génétiques qui entraînent la transcription de protéines aberrantes et l’accumulation de matériaux insolubles dans la cornée. Les troubles peuvent affecter ou non la vision et peuvent être symétriques ou non (1). Le système de classification 2015 de l’International Committee for Classification of Corneal Dystrophies (IC3D) a divisé les dystrophies cornéennes en 4 catégories : dystrophies épithéliales et sous-épithéliales, dystrophies épithéliales-stromales, dystrophies stromales et dystrophies endothéliales. La plupart des dystrophies précédemment considérées comme stromales sont maintenant classées soit comme dystrophies épithéliales-stromales, soit comme dystrophies stromales. Les tableaux 1 et 2 dressent la liste des dystrophies épithéliales-stromales et des dystrophies stromales (2). L’ancienne classification des dystrophies stromales cornéennes figure dans le tableau 3.
- Dystrophie cornéenne de Reis-Bucklers
- Dystrophie cornéenne de Thiel-Behnke
- Dystrophie cornéenne en réseau, type 1 et variantes
- Dystrophie cornéenne granuleuse, type 1
- Dystrophie cornéenne granuleuse, type 2
- Dystrophie maculaire de la cornée
- Dystrophie cornéenne de Schnyder
- Dystrophie stromale congénitale de la cornée
- Dystrophie amorphe postérieure de la cornée
- Dystrophie nuageuse centrale de François
- Dystrophie cornéenne pré-…
- .Descemet
Tableau 3. Ancienne classification des dystrophies du stroma cornéen
- Dystrophie cornéenne en réseau
- Dystrophie cornéenne granuleuse
- Dystrophie cornéenne d’Avellino
- Dystrophie cornéenne maculaire
- Dystrophie gélatineuse en gouttes
- Dystrophie cornéenne de l’âge d’or
- .gélatineuse
- Dystrophie cornéenne de Schnyder
- Dystrophie François-Neetans Fleck dystrophie
- Dystrophie stromale héréditaire congénitale
Dystrophies épithéliales-stromales cornéennes
Les dystrophies épithéliales-stromales sont causées par des mutations du gène du facteur de croissance transformant bêta (TGFβI), également connu sous le nom de gène BIGH3. Le TGFβI est situé sur le chromosome 5q31 et code pour la kératoépithéline, une protéine sécrétée par l’épithélium cornéen. Cette protéine agit comme une protéine d’adhésion et est présente dans le stroma normal. Étant une petite protéine de la taille de l’albumine, elle a la capacité de se diffuser dans le stroma cornéen. Lorsqu’une mutation du gène TGFβI se produit, la structure de la kératoépithéline est anormale et une accumulation de la protéine insoluble ou de ses fragments protéolytiques se produit dans la cornée (1, 3). Il est intéressant de noter que la mutation du gène TGFβI a été découverte en partie à l’Université de l’Iowa. Un groupe de chercheurs et de cliniciens, dont Edwin M. Stone, Robert Folberg et Jay H. Krachmer, a cartographié la dystrophie granulaire de type I, la dystrophie granulaire de type II et la dystrophie en réseau sur le chromosome 5q en 1994 (4). À ce jour, 63 mutations différentes ont été identifiées dans le gène TGFβI. Aucun traitement efficace pour prévenir ou atténuer le dépôt de la kératoépithéline n’a été identifié. Les dystrophies ont typiquement une hérédité autosomique dominante et impliquent la couche de Bowman et le stroma (3).
DYSTROPHIE CORnéENNE DE REIS-BUCKLERS
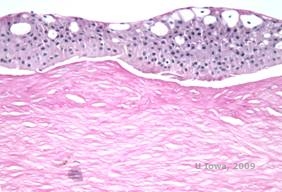
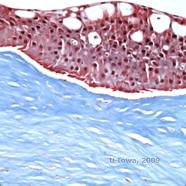
Reis-Bücklers, anciennement connue sous le nom de dystrophie cornéenne granuleuse de type III ou dystrophie cornéenne de type I de Bowman, présente typiquement des cornées normales à la naissance mais développe des érosions récurrentes douloureuses, une opacification et une perte de vision progressive dans la première décennie de vie (1). Des opacités irrégulières, gris-blanc, de type géographique, sont situées dans la couche de Bowman et le stroma antérieur. Dans les stades plus avancés de la maladie, les opacités peuvent s’étendre au limbe et au stroma plus profond (2). L’histopathologie révèle des dépôts stromaux et sous-épithéliaux antérieurs d’un matériau de type hyalin qui perturbent et souvent remplacent la couche de Bowman (voir figures 1A et 1B). Les dépôts sont colorés en rouge par la coloration au trichrome de Masson (2). Le matériau d’aspect hyalin est constitué de corps en forme de bâtonnets au niveau ultrastructurel, ce qui permet de le distinguer de la dystrophie cornéenne de Thiel-Behnke (1, 2).
| A : H&E de Reis Bückler montrant la destruction de la couche de Bowman et un épithélium irrégulier | B. Coloration de Masson Trichrome démontrant une coloration épithéliale |
 |
 |
DYSTROPHIE CORNÉENNE EN RÉSEAU
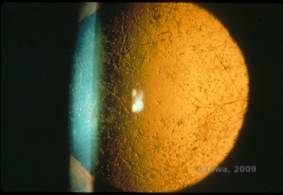
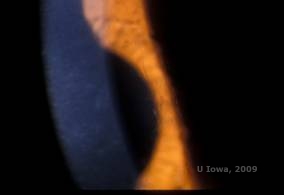
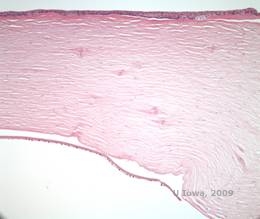
La dystrophie cornéenne en réseau (LCD) est la plus commune des dystrophies épithéliales-stromales cornéennes. C’est typiquement une maladie autosomique dominante, bilatérale, qui se présente vers la fin de la première décennie de vie avec des symptômes d’érosions cornéennes récurrentes et de baisse de la vision. Elle se caractérise par des lignes en treillis qui sont des opacités réfractiles linéaires, orientées radialement et ramifiées, décrites comme « ressemblant à du verre », situées dans le stroma antérieur (voir figures 2A et 2B). Ces lignes en treillis se trouvent initialement dans la cornée centrale superficielle. Au fur et à mesure que la maladie progresse, elles s’étendent plus profondément et en périphérie du stroma, en épargnant le limbe (1, 2). Les autres résultats de l’examen comprennent des opacités en forme de mouchetures, des points blancs sous-épithéliaux et un voile stromal « en verre dépoli », qui commence au centre et devient plus diffus (2). De nombreux patients atteints de LCD devront subir une intervention chirurgicale pour traiter les érosions récurrentes et la baisse de la vision. Si la maladie est située dans la partie antérieure du stroma, les patients peuvent souvent être traités avec succès par kératectomie photothérapeutique (PTK). Certains nécessitent une transplantation de cornée. Comme la kératoépithéline, la protéine produite par le gène TGFβI, est produite principalement dans l’épithélium cornéen, la maladie a tendance à récidiver dans les greffes de cornée (1).
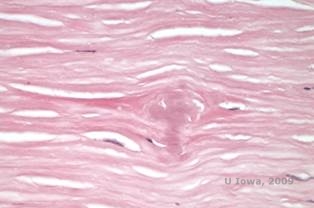
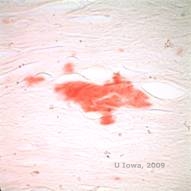
Dans la LCD, les dépôts amyloïdes s’accumulent entre la membrane basale épithéliale et la couche de Bowman ainsi que dans le stroma, provoquant une distorsion de l’architecture lamellaire. Les dépôts se colorent positivement en immunohistochimie à l’aide d’anticorps contre la kératoépithéline (2). Les dépôts apparaissent comme des dépôts roses amorphes sur la coloration à l’hématoxyline et à l’éosine (H&E) (voir Figure 1C et 1D) et se colorent avec la coloration au rouge Congo démontrant la biréfringence classique vert pomme en polarisation croisée (voir Figure 2E et 2F) (1). L’absence ou l’amincissement de la couche de Bowman, l’atrophie épithéliale et la dégénérescence de l’épithélium basal peuvent également être constatés à l’histopathologie dans la LCD (2).
L’ACL de type I est la forme classique d’ACL causée par une mutation du gène TGFβI entraînant un dépôt amyloïde isolé dans la cornée. Quatre variantes de LCD ont été identifiées : ACL de type IIIA, de type I/IIIA, de type IV et amyloïdose polymorphe. Les variantes de l’ACL se manifestent plus tard dans la vie que l’ACL classique. L’ACL de type IIIA se présente dans la 5e-7e décennie, généralement avec des érosions épithéliales. Il présente des lignes de réseau plus épaisses, décrites comme « d’apparence rugueuse », qui s’étendent jusqu’au limbe. L’ACL de type I/IIIA présente des lignes en treillis fines. L’ACL de type IV se présente dans la 7e-9e décennie avec de petites lignes en réseau. Les dépôts amyloïdes de l’ACL de type IV se trouvent dans le stroma profond et les érosions épithéliales sont rares. Les lignes de treillis sont absentes dans le type d’amylose polymorphe et les érosions épithéliales sont rares (2).
LCD type II est un syndrome d’amylose systémique connu sous le nom de syndrome de Meretoja affectant la peau, les nerfs crâniens et la cornée. Il se présente au début de l’âge adulte avec des neuropathies périphériques, des neuropathies crâniennes, un faciès de chien de chasse, une peau sèche, un blépharochalasis, des lèvres saillantes et des lignes de réseau cornéen. Ce type a été associé au gène de la gelsoline sur le chromosome 9, qui code pour une protéine précurseur de l’amyloïde dont la fonction est d’éliminer l’actine des sites de blessure et d’inflammation (1). Ce nom est une erreur d’appellation et n’est pas considéré comme une variante de la dystrophie cornéenne en treillis (2).
| A : œil gauche en rétroillumination démontrant des dépôts stromaux antérieurs dans la dystrophie cornéenne en treillis | B : œil gauche avec une puissance plus élevée montrant des dépôts stromaux antérieurs linéaires. |
 |
 |
| C : Coloration H&E de la cornée en treillis. Notez les dépôts amorphes roses dans le stroma | D : Vue rapprochée des dépôts amorphes roses |
 |
 |
| E : Coloration au rouge Congo, mettant en évidence l’amyloïde | F : Biréfringence vert pomme de l’amyloïde avec polarisation croisée. |
 |
 |
DYSTROPHIE CORNÉALE GRANULAIRE, TYPE I
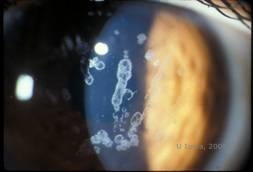
Dystrophie cornéenne granulaire, type I (GCD1) est une maladie bilatérale, autosomique dominante, associée à une mutation du gène TGFβI qui entraîne le dépôt d’un matériau hyalin dans le stroma cornéen. Elle se manifeste typiquement au début de la première décennie de vie par des opacités gris-blanc, » en forme de miettes « , dans le stroma antérieur à moyen, s’étendant dans le stroma postérieur en cas de maladie avancée (1, 2). Ces opacités sont des dépôts discrets situés au centre, avec une cornée claire en périphérie et une cornée claire entre les dépôts (voir figures 3A et 3B). La maladie est généralement asymptomatique au début, mais avec le temps, les opacités peuvent coalescer et entraîner une baisse de la vision. Des érosions cornéennes récurrentes peuvent se produire dans le cas du GCD, mais à une incidence plus faible que dans le cas du LCD (1, 5). Les patients peuvent également souffrir d’éblouissement et de photophobie (2). Au début du processus pathologique, le traitement se limite souvent à l’observation. Cependant, à mesure que la maladie progresse, une PTK et une transplantation de cornée peuvent être nécessaires pour améliorer la vision et éroder les symptômes. Comme la LCD, la maladie peut récidiver dans les greffes de cornée.
Histopathologiquement, les opacités sont des dépôts éosinophiles souvent décrits comme « ressemblant à des bonbons de roche » dans le stroma antérieur, composés d’un matériau de type hyalin. Avec le temps, les dépôts progressent dans le stroma cornéen plus profond. Le matériau hyalin se colore en rouge vif avec la coloration au trichrome de Masson (voir figure 3C et 3D).
| A:Photo à la lampe à fente de la dystrophie cornéenne granuleuse, type I | B : Notez les dépôts stromaux « friables » avec un stroma clair intermédiaire. |
 |
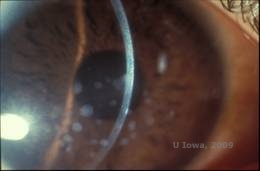 |
| C : Coloration H& E de la cornée montrant des dépôts hyalins éosinophiles « ressemblant à du sucre candi » dans le stroma | D : Le matériel hyalin se colore en rouge vif avec Masson-Trichrome |
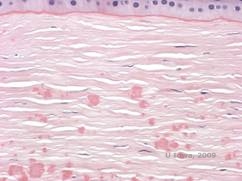 |
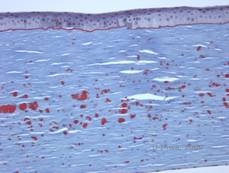 |
DYSTROPHIE CORNÉENNE GRANULAIRE, TYPE II
Dystrophie cornéenne granulaire, type II (GCD2), anciennement appelée dystrophie cornéenne d’Avellino ou combinée à un réseau granulaire, est une maladie autosomique dominante liée à une mutation du gène TGFβI qui entraîne un dépôt à la fois hyalin et amyloïde dans le stroma cornéen. Généralement, les patients se présentent dans la deuxième décennie de leur vie avec de petits points gris-blancs dans le stroma superficiel. Les opacités peuvent également avoir une forme d’épine, d’anneau ou d’étoile. En rétroillumination, elles sont partiellement translucides. Plus tard dans le processus de la maladie, elles peuvent également développer des lignes en réseau (voir figures 4A et 4B). Ces lignes ne se croisent pas et apparaissent plus blanches et moins réfringentes que les lignes en treillis. Les symptômes du GCD2 sont des douleurs accompagnées d’érosions épithéliales et d’une déficience visuelle (2).
Au niveau histopathologique, la cornée présentera des dépôts stromaux qui se colorent en rouge avec le Trichrome de Masson, indiquant la présence de hyaline (Voir Figure 4C). En outre, la coloration au rouge Congo montre une biréfringence vert pomme en polarisation croisée, indiquant la présence d’amyloïde (voir figure 4D). On pensait que la maladie était originaire d’une famille d’Avellino, en Italie. Cependant, la DCG de type II a maintenant été rapportée chez des patients de nombreux autres pays également (2,5), la prévalence la plus élevée étant en Asie de l’Est.
| A : Dystrophie d’Avellino montrant des dépôts de type treillis et de type granulaire dans le stroma cornéen | B. Tache de Masson Trichrome démontrant des dépôts hyalins stromaux antérieurs |
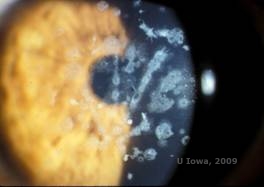 |
 |
| C : Tache de rouge Congo montrant des dépôts amyloïdes roses et amorphes dans le même spécimen cornéen. | D : La polarisation croisée révèle une biréfringence vert pomme indiquant une amyloïde. |
 |
 |
DYSTROPHIES CORNÉALES STROMALES
DYSTROPHIE CORNÉALE MACULAIRE
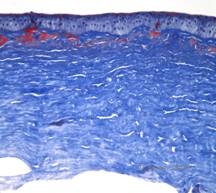
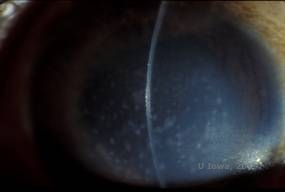
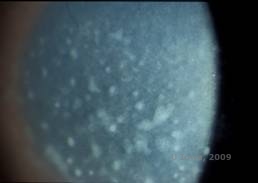
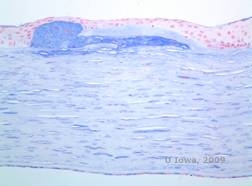
Dystrophie cornéenne maculaire. (MCD) est une maladie autosomique récessive causée par une mutation du gène de la sulfotransférase 6 des glucides (CHST6) sur le chromosome 16 qui entraîne un défaut de synthèse du sulfate de kératan, le principal glycosaminoglycane de la cornée. Elle est moins fréquente que la LCD ou la GCD, mais tend à avoir un impact plus sévère sur la vision. Bien que la MCD soit moins fréquente dans le monde que la LCD ou la GCD, elle est la plus courante des dystrophies stromales cornéennes dans des pays comme l’Islande et l’Arabie saoudite (2,6). Des lésions stromales antérieures de couleur gris-blanc et ressemblant à des mouchetures, semblables à celles de la GCD1, apparaissent dans la cornée au cours de la première décennie de la vie. Cependant, contrairement au GCD1, il existe un voile stromal entre les dépôts et toute la cornée, du limbe au limbe, est souvent touchée (voir figures 5A et 5B). La cornée est fine et, au fur et à mesure que la maladie progresse, la membrane de Descemet devient grise et développe des guttae. Des érosions épithéliales peuvent se produire, mais moins dans le MCD que dans le LCD. Les patients développent généralement une perte visuelle sévère entre la deuxième et la troisième décennie de leur vie en raison d’un voile cornéen diffus. La PTK peut être réalisée dans certains cas précoces de MCD. Cependant, cette affection ne se prête généralement pas autant à la PTK que la dystrophie en réseau ou la dystrophie granulaire et nécessite souvent une greffe de cornée pour être traitée (7). La récidive dans les greffes est moins fréquente dans la MCD que dans la dystrophie granulaire ou en réseau (1,2,5,6,8).
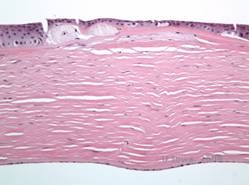
Les dépôts stromaux dans la MCD sont composés de mucopolysaccharides qui s’accumulent dans le réticulum endoplasmique des kératocytes du stroma cornéen, de manière extracellulaire entre les lamelles stromales, et dans l’épithélium, la membrane de Descemet et l’endothélium. Ces dépôts se colorent en bleu avec le bleu Alcian (voir figure 5C et 5D) (1). On observe des ruptures de la couche de Bowman et des guttae avec un épaississement de la membrane de Descemet (2).
Trois sous-types de MCD ont été décrits en fonction de la présence ou de l’absence de sulfate de kératan immunoréactif au sein de divers tissus. Le type I ne présente pas de kératan sulfate immunoréactif dans le stroma cornéen, les kératocytes, le sérum ou le cartilage, et constitue la variante la plus courante de MCD dans le monde. Le type IA est dépourvu de sulfate de kératan dans le stroma, le sérum et le cartilage, mais présente des niveaux détectables dans les kératocytes. Le type II a du sulfate de kératan présent à des niveaux très réduits dans le stroma, les kératocytes, les sérums et le cartilage (6).
| A : photo à la lampe à fente de la dystrophie cornéenne maculaire. | B : Notez le voile entre les dépôts stromaux cornéens |
 |
 |
| C : H&E de cornée avec dystrophie maculaire. Notez les dépôts stromaux antérieurs et la perturbation de la couche de Bowman | D : Dépôts de mucopolysaccharides au sein des kératocytes mis en évidence par la coloration au bleu Alcian |
 |
 |
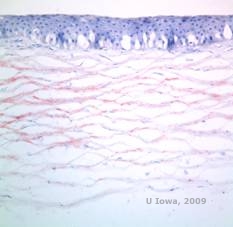
DYSTROPHIE CORnéENNE DE SCHNYDER (DSC)
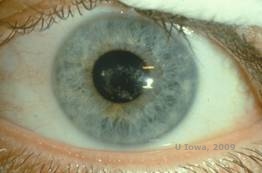
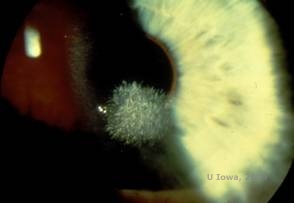
Dystrophie cornéenne de Schyder (DSC), précédemment connue sous le nom de dystrophie cornéenne cristalline de Schnyder, est une dystrophie stromale cornéenne bilatérale autosomique dominante liée à une mutation génétique du gène UbiA prenyltransferase domain containing 1(UBIAD1) sur le chromosome 1. Le défaut métabolique des kératocytes cornéens qui en résulte entraîne un dépôt de cholestérol cristallin dans le stroma. Cependant, la présence de cristaux n’est pas absolument nécessaire pour le diagnostic du SCD. En fait, seuls 54 % des patients atteints de DSC présentent des cristaux cornéens. En général, les patients se présentent dans la deuxième ou troisième décennie avec une opacité cornéenne centrale en forme d’anneau avec ou sans cristaux sous-épithéliaux en forme de virgule (voir figures 6A et 6B). Ensuite, l’arcus lipoides apparaît entre 23 et 38 ans. Après l’âge de 38 ans, l’opacification progressive de la cornée se traduit par un voile panstromal atteignant la mi-périphérie. La plupart des patients âgés de plus de 50 ans présentent une perte de vision photopique, un éblouissement et une diminution de la sensation cornéenne, et peuvent donc nécessiter un traitement chirurgical comprenant une greffe de cornée ou une PTK. Une récidive dans la greffe peut se produire. La maladie a été associée à une hypercholestérolémie, une hyperlipidémie et un genu valgum chez certains patients (2,5,9,10).
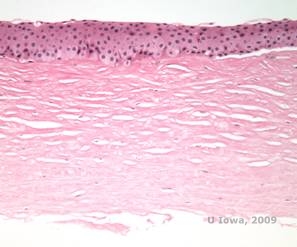
Histopathologiquement, des cristaux de cholestérol biréfringents composés de phospholipides et de cholestérol se déposent à l’intérieur des cellules épithéliales basales, des kératocytes, de la couche de Bowman et entre les lamelles stromales. Les lipides se dissolvent dans le traitement histologique normal, de sorte que des sections congelées à travers la cornée doivent être obtenues pour démontrer la présence de lipides avec des colorations Oil-Red-O ou noir Soudan.
| A : photo à la lampe à fente de la dystrophie cornéenne de Schnyder. | B. Dépôts cristallins situés au centre |
 |
 |
| C : H&E de la cornée avec SCCD | D. La coloration Oil Red O met en évidence les cristaux de cholestérol qui apparaissent en rouge. |
 |
 |
Le tableau 4 fournit un mnémonique commun pour mémoriser certaines des dystrophies cornéennes affectant le stroma, la composition de leur dépôt, et la méthode de coloration de ces dépôts est listée.
Tableau 4 : Mnémonique pour se souvenir des dystrophies du stroma cornéen
- Dystrophie de Marilyn-Maculaire
- Monroe-Mucopolysaccharide
- Coloration au bleu d’Alcian toujours
- Gets-Granulaire. Dystrophie
- Her-Hyaline
- Tache Man in-Masson Trichrome
- Dystrophie Los-Lattice
- Angeles-Amyloïde
- California-Congo Red
VISUALISATION : DYSTROPHIES CORNEALES STROMALES
EPIDEMIOLOGIE
|
SIGNES
|
SYMPTÔMES
|
TRAITEMENT
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Pathologie conjonctivale et cornéenne. Principes et pratique de l’ophtalmologie d’Albert et Jakobiec, 3e édition. Saunders. 2008 : 3592-3595.
- Weiss JS, et al. Classification IC3D des dystrophies cornéennes–édition 2. Cornea 2015 ; 34 (2) : 117-159.
- Lakshminarayanan R, et al. Aspects cliniques et génétiques des dystrophies cornéennes associées au TGFBI. Ocul Surf 2014 ; 12 (4) : 234-251.
- Stone EM, et al. Trois dystrophies cornéennes autosomiques dominantes se cartographient sur le chromosome 5q. Nature genet. 1994 ; 6 : 47-51.
- Bron AJ. Génétique des dystrophies cornéennes : Ce que nous avons appris au cours des vingt-cinq dernières années. Cornea 2000 ; 19(5) : 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Kératoplastie pénétrante pour la dystrophie cornéenne maculaire. Ophthalmology 2005;112 : 220-224.
- Badr IA, Wagoner MD. Kératectomie photothérapeutique pour la dystrophie cornéenne maculaire. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Génétique des dystrophies cornéennes antérieures et stromales. Seminars in Ophthalmology, 2008 ; 23 : 1,9-17.
- Weiss JS. Dystrophie cornéenne de Schnyder. Curr Opin Ophthalmol 2009 ; 20 (4) : 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. Le gène de la dystrophie cornéenne cristalline de Schnyder est cartographié sur le chromosome humain 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Format de citation suggéré : Van C, Syed NA. Dystrophies cornéennes épithéliales-stromales et stromales : A Clinicopathologic Review. Révision de ; EyeRounds.org. 20 août 2015. Disponible sur : http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm