- En klinisk-patologisk gennemgang
- INDLEDNING
- EPITHELIAL-STROMAL CORNEAL DYSTROPHIES
- REIS-BUCKLERS CORNEAL DYSTROPHY
- LATTICE CORNEAL DYSTROPHY
- GRANULAR CORNEAL DYSTROPHY, TYPE I
- GRANULAR CORNEAL DYSTROPHY, TYPE II
- STROMAL CORNEAL DYSTROPHIES
- MACULAR CORNEAL DYSTROPHY
- SCHNYDER CORNEAL DYSTROPHY (SCD)
- Tabel 4: Mnemoteknik til at huske corneale stromale dystrophier
- OVERVIEW: CORNEAL STROMAL DYSTROPHIES
- EPIDEMIOLOGI
- SIGNER
- SYMPTOMER
- BEHANDLING
En klinisk-patologisk gennemgang
Emily S. Birkholz, MD, Nasreen A. Syed, MD, og Michael D. Wagoner, MD, PhD
August 17, 2009
Større revision: Chaunhi Van, MD og Nasreen Syed, MD
20. august 2015
INDLEDNING
Corneale epithel-stromale og stromale dystrophier er en gruppe af arvelige sygdomme i cornea, der skyldes en progressiv ophobning af aflejringer i corneaens lag. Disse aflejringer er ikke forårsaget af inflammation, infektion eller traumer, men af genetiske mutationer, der fører til transkription af afvigende proteiner, hvilket resulterer i en ophobning af uopløseligt materiale i hornhinden. Disse sygdomme kan påvirke synet eller ej og kan være symmetriske eller ej (1). Det internationale klassifikationssystem IC3D (International Committee for Classification of Corneal Dystrophies) fra 2015 har inddelt hornhindedystrophier i 4 kategorier: epithel- og subepitheldystrophier, epithel-strømsdystrophier, stromale dystrophier og endotheldystrophier. De fleste dystrophier, der tidligere blev betragtet som stromale, klassificeres nu som enten epithelial-stromale dystrophier eller stromale dystrophier. Tabel 1 og 2 indeholder en liste over epithelial-stromale dystrophier og stromale dystrophier (2). Den gamle klassifikation for corneale stromale dystrophier er anført i tabel 3.
- Reis-Bucklers corneadystrofi
- Thiel-Behnke corneadystrofi
- Lattice corneadystrofi, type 1 og varianter
- Granulær hornhindedystrofi, type 1
- Granulær hornhindedystrofi, type 2
- Makulær hornhindedystrofi
- Schnyder-hornhindedystrofi
- Medfødt stromal hornhindedystrofi
- Posterior amorf hornhindedystrofi
- Central cloudy dystrophy of Francois
- Pre-Descemet corneal dystrophy
Tabel 3. Gammel klassifikation af corneale stromale dystrophier
- Lattice corneal dystrophy
- Granulær corneal dystrophy
- Avellino corneal dystrophy
- Makulær corneal dystrophy
- Gelatinøs dråbe-lignende dystrofi
- Schnyder corneal dystrofi
- Francois-Neetans Fleck dystrofi
- Medfødt arvelig stromal dystrofi
EPITHELIAL-STROMAL CORNEAL DYSTROPHIES
Epithelial-stromal dystrophies er forårsaget af mutationer i transforming growth factor beta-induceret (TGFβI) genet, også kendt som BIGH3-genet. TGFβI er placeret på kromosom 5q31 og koder for keratoepithelin, et protein, der udskilles af hornhindeepitelet. Dette protein fungerer som et adhæsionsprotein og er til stede i normalt stroma. Da det er et lille protein på nogenlunde samme størrelse som albumin, har det mulighed for at diffundere gennem det corneale stroma. Når der opstår en mutation i TGFβI-genet, er keratoepithelinstrukturen unormal, og der sker en ophobning af det uopløselige protein eller dets proteolytiske fragmenter i cornea (1, 3). Interessant nok blev TGFβI-genmutationen delvis opdaget på University of Iowa. En gruppe af forskere og klinikere, herunder Edwin M. Stone, Robert Folberg og Jay H. Krachmer, kortlagde granulær type I, granulær type II og gitterdystrofi på kromosom 5q i 1994 (4). Til dato er der blevet identificeret 63 forskellige mutationer i TGFβI-genet. Der er ikke blevet identificeret nogen effektiv behandling til at forhindre eller dæmpe aflejringen af keratoepithelin. Dystroferne har typisk en autosomal dominant arvelighed og involverer Bowman-laget og stroma (3).
REIS-BUCKLERS CORNEAL DYSTROPHY
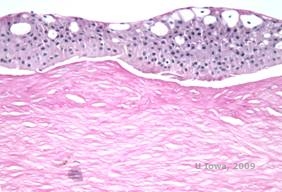
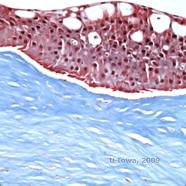
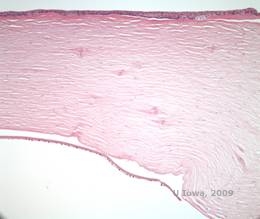
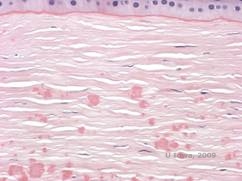
Reis-Bücklers, tidligere kendt som Granular corneal dystrophy type III eller Corneal Dystrophy of Bowman’s type I, præsenterer sig typisk med normale hornhinder ved fødslen, men udvikler smertefulde tilbagevendende erosioner, opacificering og progressivt synstab inden for det første årti af livet (1). Uregelmæssige, gråhvide, geografisk lignende opaciteter er lokaliseret i Bowman-laget og det forreste stroma. I mere fremskredne stadier af sygdommen kan opaciteterne strække sig til limbus og det dybere stroma (2). Histopatologien afslører anteriore stromale og subepitheliale aflejringer af hyalinlignende materiale, som afbryder og ofte erstatter Bowman-laget (se figur 1A og 1B). Aflejringerne farves rødt med Masson-trichromfarve (2). Det hyalinlignende materiale består ultrastrukturelt af stavlignende organer, hvilket er med til at skelne det fra Thiel-Behnke corneal dystrofi (1, 2).
| A: H&E af Reis Bückler, der viser ødelæggelse af Bowmans lag og uregelmæssigt epitel | B. Masson Trichrome farvning, der viser epitelfarvning |
 |
 |
LATTICE CORNEAL DYSTROPHY
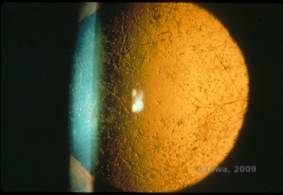
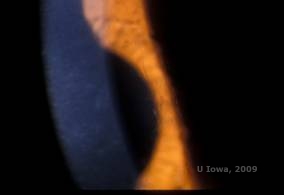
Lattice corneal dystrophy (LCD) er den mest almindelige af de corneale epithel-strømme dystrophier. Det er typisk en autosomalt dominerende, bilateral sygdom, der typisk viser sig mod slutningen af det første årti af livet med symptomer på tilbagevendende hornhindeerosioner og nedsat syn. Den er karakteriseret ved gitterlinjer, som er lineære, radialt orienterede, forgrenede refraktile opaciteter, der beskrives som “glasagtige” og er placeret i det forreste stroma (se figur 2A og 2B). Disse gitterlinjer findes i første omgang i den overfladiske centrale cornea. Efterhånden som sygdommen skrider frem, spredes de dybere og perifert i stromaet med skånelse af limbussen (1, 2). Andre undersøgelsesfund omfatter fleck-lignende opaciteter, subepitheliale hvide prikker og “ground-glass” stromal tåge, som starter centralt og bliver mere diffus (2). Mange patienter med LCD vil kræve kirurgisk indgreb til behandling af tilbagevendende erosioner og nedsat syn. Hvis sygdommen er lokaliseret fortil i stromaet, kan patienterne ofte behandles med succes med fototerapeutisk keratektomi (PTK). Nogle kræver hornhindetransplantation. Da keratoepithelin, det protein, der produceres af TGFβI-genet, hovedsageligt produceres i hornhindeepitelet, har sygdommen tendens til at vende tilbage i hornhindetransplantater (1).
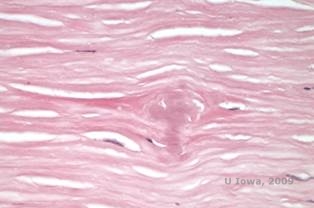
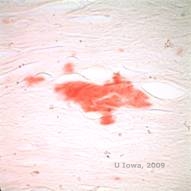
Ved LCD ophobes amyloidaflejringer mellem den epitheliale basalmembran og Bowman-laget samt i stromaet, hvilket forårsager forvrængning af den lamellære arkitektur. Aflejringerne farves positivt med immunohistokemi ved hjælp af antistoffer mod keratoepithelin (2). Aflejringerne fremstår som amorfe lyserøde aflejringer på hæmatoxylin- og eosin (H&E)-farve (se figur 1C og 1D) og farves med Kongorødfarve, der viser den klassiske æblegrønne dobbeltring ved krydspolarisering (se figur 2E og 2F) (1). Manglende eller udtynding af Bowman-laget, epithelatrofi og basal epitheldegeneration kan også findes på histopatologi i LCD (2).
LCD type I er den klassiske form for LCD forårsaget af en mutation i TGFβI-genet, der resulterer i isoleret amyloidaflejring i cornea. Der var blevet identificeret fire LCD-varianter: LCD type IIIA, type I/IIIA, type IV og polymorfe amyloidose. LCD-varianter viser sig senere i livet end klassisk LCD. LCD type IIIA viser sig i det 5.-7. årti, sædvanligvis med epithelerosioner. Den har tykkere gitterlinjer, der beskrives som “ropy-lignende”, og som strækker sig til limbussen. LCD type I/IIIA har tynde gitterlinjer. LCD type IV viser sig i det 7.-9. årti med små gitterlinjer. Amyloidaflejringer i LCD type IV findes i det dybe stroma, og der forekommer sjældent epithelerosioner. Gitterlinjer er fraværende i polymorf amyloidose type, og der forekommer sjældent epitelerosioner (2).
LCD type II er et systemisk amyloidosesyndrom kendt som Meretoja syndrom, der påvirker hud, kranienerver og cornea. Det viser sig i tidlig voksenalder med perifere neuropatier, kraniale neuropatier, hundelignende ansigtsudtryk, tør hud, blepharochalasis, fremspringende læber og hornhindegitterlinjer. Denne type er blevet knyttet til gelsolin-genet på kromosom 9, som koder for et amyloidprækursorprotein, der fungerer til at fjerne actin fra skadesteder og betændelsessteder (1). Navnet er en misvisende betegnelse og anses ikke for at være en variant af gitterhindehindedystrofi (2).
| A: Venstre øje ved retroilluminering, der viser anteriore stromale aflejringer ved lattice corneal dystrophy | B: Venstre øje med højere effekt, der viser lineære anteriore stromale aflejringer. |
 |
 |
| C: H&E farvning af cornea med gitter. Bemærk lyserøde amorfe aflejringer i stroma | D: Nærmere billede af de lyserøde, amorfe aflejringer |
 |
 |
| E: Kongorød farvning, der fremhæver amyloid | F: Æblegrøn dobbeltbrydning af amyloid med krydspolarisering. |
 |
 |
GRANULAR CORNEAL DYSTROPHY, TYPE I
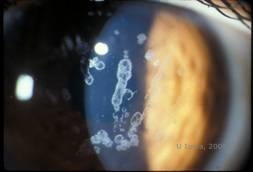
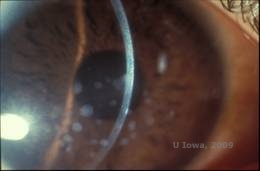
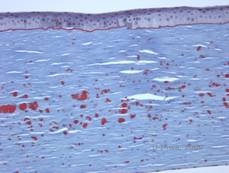
Granulær hornhindedystrofi, type I (GCD1) er en bilateral, autosomal dominant sygdom, der er forbundet med en mutation i TGFβI-genet, som fører til aflejring af et hyalinmateriale i det corneale stroma. Den viser sig typisk tidligt i det første årti af livet med grå-hvide, “krumme-lignende” opaciteter i det forreste til midterste stroma, som strækker sig ind i det bageste stroma ved fremskreden sygdom (1, 2). Disse opaciteter er diskrete aflejringer placeret centralt, med klar cornea placeret i periferien og klar cornea mellem aflejringerne (se figur 3A og 3B). Sygdommen er typisk asymptomatisk på et tidligt tidspunkt, men med tiden kan opaciteterne vokse sammen og føre til nedsat syn. Recidiverende corneaerosioner kan forekomme ved GCD, men med en lavere forekomst end ved LCD (1, 5). Patienterne kan også opleve blænding og fotofobi (2). Behandlingen på et tidligt tidspunkt i sygdomsforløbet er ofte kun observation. Efterhånden som sygdommen udvikler sig, kan det imidlertid være nødvendigt med PTK og hornhindetransplantation for at forbedre synet og erosionssymptomerne. Ligesom LCD kan sygdommen recidivere i corneatransplantationer.
Histopatologisk er opaciteterne eosinofile aflejringer, der ofte beskrives som “rock candy-agtige” i det forreste stroma lavet af et hyalin-lignende materiale. Med tiden udvikler aflejringerne sig ind i det dybere hornhindestroma. Det hyaline materiale farves knaldrødt med Masson trichrome farvestof (se figur 3C og 3D).
| A:Spaltlampefoto af granulær hornhindedystrofi, type I | B: Bemærk de “krummelignende” stromale aflejringer med klart mellemliggende stroma. |
 |
 |
| C: H& E farvning af cornea, der viser eosinofile “klippekrummelignende” hyaline aflejringer i stroma | D: Hyalinmateriale farves klart rødt med Masson-Trichrome |
 |
 |
GRANULAR CORNEAL DYSTROPHY, TYPE II
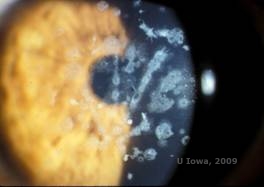
Granular corneal dystrophy, type II (GCD2), tidligere kendt som Avellino eller kombineret granulær-net hornhindedystrofi, er en autosomal dominant sygdom, der er knyttet til en mutation i TGFβI-genet, som fører til en aflejring af både hyalin og amyloid i hornhindestromaet. Typisk viser patienterne sig i det andet årti i livet med små gråhvide prikker i det overfladiske stroma. Opaciteterne kan også være tornlignende, ringformede eller stjerneformede. Ved retroillumination er de delvist gennemsigtige. Senere i sygdomsforløbet kan de også udvikle gitterlinjer (se figur 4A og 4B). Disse linjer krydser ikke hinanden og fremstår hvidere og mindre brydbare end gitterlinjer. Symptomer på GCD2 er smerter med epithelerosioner og synsforstyrrelser (2).
Histopatologisk vil hornhinden have stromale aflejringer, der farves rødt med Masson Trichrome, hvilket indikerer tilstedeværelsen af hyalin (Se figur 4C). Desuden vil farvning med congorødt vise æblegrøn dobbeltfringence ved krydspolarisering, hvilket indikerer tilstedeværelsen af amyloid (se figur 4D). Sygdommen menes at stamme fra en familie i Avellino, Italien. GCD type II er imidlertid nu også blevet rapporteret hos patienter fra mange andre lande (2,5), og den højeste prævalens findes i Østasien.
| A: Avellino dystrofi, der viser gitterlignende og granulære lignende aflejringer i hornhindestroma | B. Masson Trichrome farvning, der viser forreste stromale hyalinaflejringer |
 |
 |
| C: Kongorød farvning, der viser lyserøde, amorfe amyloide aflejringer i samme hornhindeprøve. | D: Krydspolarisering afslører æblegrøn dobbeltfringence, der indikerer amyloid. |
 |
 |
STROMAL CORNEAL DYSTROPHIES
MACULAR CORNEAL DYSTROPHY
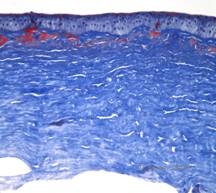
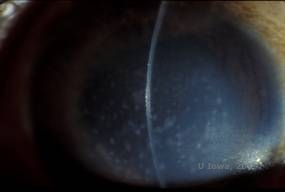
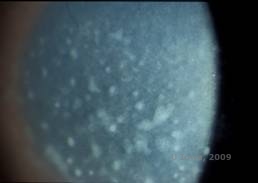
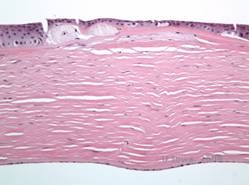
Maculær corneal dystrofi (MCD) er en autosomal recessiv sygdom forårsaget af en mutation i genet for kulhydratsulfotransferase 6 (CHST6) på kromosom 16, som fører til en defekt i syntesen af keratansulfat, det vigtigste glykosaminoglykan i hornhinden. Den er mindre almindelig end LCD eller GCD, men den har tendens til at påvirke synet mere alvorligt. Selv om MCD er mindre almindelig på verdensplan end LCD eller GCD, er det den mest almindelige af de corneale stromale dystrophier i lande som Island og Saudi-Arabien (2,6). Grå-hvide, fleksagtige forreste stromale læsioner, der ligner GCD1, forekommer i hornhinden i det første årti af livet. I modsætning til GCD1 er der imidlertid stromal uklarhed mellem aflejringerne, og hele cornea fra limbus til limbus er ofte involveret (se figur 5A og 5B). Cornea er tynd, og efterhånden som sygdommen skrider frem, bliver Descemetmembranen grå og udvikler guttae. Der kan forekomme epithelerosioner, men i mindre grad i MCD end i LCD. Patienterne udvikler typisk et alvorligt synstab i det andet til tredje årti af deres liv på grund af diffus hornhindeudsløring. PTK kan udføres i nogle tidlige tilfælde af MCD. Denne tilstand er dog generelt ikke så velegnet til PTK som gitter- eller granulær dystrofi og kræver ofte hornhindetransplantation til behandling (7). Recidiv i transplantater er mindre almindelig ved MCD end ved granulær eller gitterdystrofi (1,2,5,6,8).
De stromale aflejringer i MCD består af mucopolysaccharider, der ophobes i det endoplasmatiske retikulum af keratocytter i hornhindestromaet, ekstracellulært mellem stromale lameller og i epitelet, Descemet-membranen og endothelet. Disse aflejringer farves blå med Alcianblå (se figur 5C og 5D) (1). Der er brud i Bowman-laget og guttae med fortykkelse af Descemet-membranen (2).
Tre undertyper af MCD er blevet beskrevet på baggrund af tilstedeværelsen eller fraværet af immunreaktivt keratansulfat i forskellige væv. Type I har ikke immunoreaktivt keratansulfat i hornhindestromaet, keratocytter, sera eller brusk og er den mest almindelige variant af MCD på verdensplan. Type IA mangler keratansulfat i stroma, sera og brusk, men har påviselige niveauer i keratocytter. Type II har keratansulfat til stede i meget reducerede niveauer i stroma, keratocytter, sera og brusk (6).
| A: Spaltelampefoto af makulær hornhindedystrofi. | B: Bemærk sløret mellem de corneale stromale aflejringer | |
 |
 |
|
| C: H&E af cornea med makulær dystrofi. Bemærk anteriore stromale aflejringer og Bowmans lagforstyrrelser | D: Mucopolysaccharidaflejringer i keratocytter fremhævet med Alcian Blue farvning | |
 |
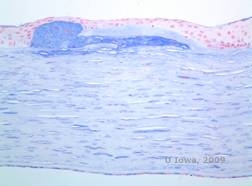 |
SCHNYDER CORNEAL DYSTROPHY (SCD)
Schnyder corneal dystrophy (SCD), tidligere kendt som Schnyder crystalline corneal dystrophy, er en autosomal dominant, bilateral corneal stromal dystrofi knyttet til en genetisk mutation i UbiA prenyltransferase domain containing 1(UBIAD1) genet på kromosom 1. Den deraf følgende metaboliske defekt i hornhindekeratocytterne fører til krystallinsk kolesterolaflejring i stromaet. Tilstedeværelsen af krystaller er dog ikke absolut nødvendig for at stille diagnosen SCD. Faktisk er det kun 54 % af patienterne med SCD, der har krystaller i hornhinden. Typisk præsenterer patienterne sig i andet eller tredje årti med en ringformet central corneal uklarhed med eller uden kommaformede subepitheliale krystaller (se figur 6A og 6B). Derefter optræder arcus lipoides mellem 23 og 38 år. Efter 38-årsalderen resulterer en progressiv corneal clouding i en panstromal tåge, der når frem til midterperiferien. De fleste patienter over 50 år har fotopisk synstab, blænding og nedsat hornhindefornemmelse, og de kan derfor kræve kirurgisk behandling, herunder hornhindetransplantation eller PTK. Der kan forekomme recidiv i transplantatet. Sygdommen er blevet forbundet med hyperkolesterolæmi, hyperlipidæmi og genu valgum hos nogle patienter (2,5,9,10).
Histopatologisk set aflejrer birefribende kolesterolkrystaller bestående af fosfolipider og kolesterol sig inden for basale epitelceller, keratocytter, Bowman’s lag og mellem stromale lameller. Lipiderne opløses ved normal histologisk behandling, så der skal udtages frosne snit gennem corneaen for at påvise tilstedeværelsen af lipid med olie-rød-O- eller sudanesortfarve.
| A: Spaltelampefoto af Schnyder corneal dystrofi. | B. Centralt placerede krystallinske aflejringer |
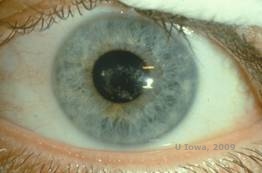 |
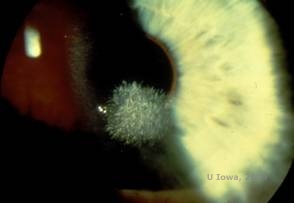 |
| C: H&E af cornea med SCCD | D. Oil Red O-farve fremhæver kolesterolkrystaller, som fremstår røde. |
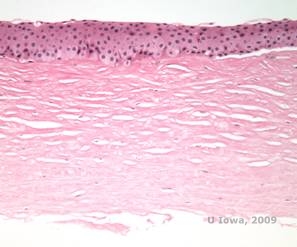 |
 |
Tabel 4 giver en fælles huskeseddel til at huske nogle af de corneale dystrophier, der påvirker stromaet, sammensætningen af deres aflejring, og metoden til farvning af disse aflejringer er anført.
Tabel 4: Mnemoteknik til at huske corneale stromale dystrophier
- Marilyn-Macular Dystrophy
- Monroe-Mucopolysaccharide
- Always-Alcian Blue stain
- Gets-Granular Dystrophy
- Her-Hyaline
- Man in-Masson Trichrome farvning
- Los-Lattice Dystrophy
- Angeles-Amyloid
- California-Congo Red
OVERVIEW: CORNEAL STROMAL DYSTROPHIES
EPIDEMIOLOGI
|
SIGNER
|
SYMPTOMER
|
BEHANDLING
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Konjunktival og corneal patologi. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3. udgave. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D klassifikation af corneale dystrophier–udgave 2. Cornea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. Kliniske og genetiske aspekter af TGFBI-associerede hornhinde-dystrophier. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nature genet. 1994; 6: 47-51.
- Bron AJ. Genetik af hornhindedystrophier: Hvad vi har lært i de sidste femogtyve år. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrerende keratoplastik til behandling af makulær hornhindedystrofi. Ophthalmology 2005;112: 220-224.
- Badr IA, Wagoner MD. Fototerapeutisk keratektomi til behandling af makulær hornhindedystrofi. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetics of anterior and stromal corneal dystrophies. Seminars in Ophthalmology, 2008; 23: 1,9-17.
- Weiss JS. Schnyder corneal dystrophy. Curr Opin Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. Genet for Schnyder’s krystallinsk hornhindedystrofi kortlægger til det menneskelige kromosom 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Foreslået citatformat: Van C, Syed NA. Epithelial-Stromal og Stromal Corneal Dystrophies: A Clinicopathologic Review. Revision af ; EyeRounds.org. August 20, 2015. Tilgængelig fra: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm