ABOVE: modified from © ISTOCK.com, tera vector
Quasi sempre, costruire qualcosa è più difficile che demolirlo. Allo stesso modo, battere i geni rappresenta una sfida maggiore che metterli fuori uso. È una realtà che i ricercatori dovranno superare per ottenere il massimo dall’editing genico. Il knock in dei geni permette agli scienziati di studiare gli effetti di specifiche varianti geniche, di usare geni reporter come la proteina fluorescente verde per tracciare i prodotti genici nel tempo e nello spazio, di sondare la regolazione del genoma e, infine, di riparare i geni che causano malattie. “È un modo davvero efficace per interrogare ogni base di un gene”, dice Greg Findlay, un candidato MD/PhD all’Università di Washington.
CRISPR-Cas9, una tecnologia di editing genico nota per la sua facilità d’uso, può mettere fuori combattimento i geni. L’eliminazione di un gene comporta l’inserimento di CRISPR-Cas9 in una cellula utilizzando un RNA guida che punta lo strumento al gene di interesse. Lì, Cas9 taglia il gene, tagliando entrambi i filamenti di DNA, e il normale meccanismo di riparazione del DNA della cellula ripara il taglio usando un processo chiamato non-homologous end joining (NHEJ). NHEJ è altamente efficiente ma impreciso. Il processo tende a introdurre errori sotto forma di piccole inserzioni o delezioni che di solito sono sufficienti per mettere fuori gioco il gene.
Per mettere fuori gioco un gene, tuttavia, i tagli devono essere riparati in modo molto preciso, senza ulteriori inserzioni o delezioni. Questo richiede lo sfruttamento di un secondo meccanismo di riparazione del DNA chiamato riparazione diretta dall’omologia (HDR), che – almeno nelle cellule dei mammiferi – avviene in modo meno efficiente, quindi la sua frequenza è inferiore a quella di NHEJ. A complicare ulteriormente il processo c’è il fatto che alcuni loci genici e tipi di cellule sono intrinsecamente meno ospitali per l’editing CRISPR-Cas9.
Negli ultimi anni, i ricercatori hanno sviluppato molte nuove strategie per aumentare l’efficienza di battere in geni grandi e piccoli usando CRISPR-Cas9, e lungo la strada hanno proposto e testato nuove applicazioni per questo tipo di editing genico. Qui, The Scientist esplora alcuni degli approcci più promettenti.
Seleziona
Ricercatore: Jon Chesnut, direttore senior della biologia sintetica R&D, Thermo Fisher Scientific
Progetto: Nello sviluppo di un kit di tagging genico chiamato Truetag che Thermo Fisher metterà sul mercato più tardi quest’anno, Chesnut ha usato marcatori selezionabili per migliorare
l’efficienza. Un marcatore selezionabile – in questo caso, un gene di resistenza agli antibiotici – è attaccato a un tag di proteina fluorescente e bussato in cellule mammifere. Queste cellule sono poi coltivate in coltura con l’antibiotico associato. Il gene di resistenza conferisce un vantaggio selettivo alle cellule che lo portano; solo loro sono in grado di crescere, e quindi quelle che crescono contengono il gene tag di interesse. Anche se l’efficienza dell’inserimento del gene è bassa, i ricercatori possono usare la selezione antibiotica per una settimana o più per finire con un’alta percentuale di cellule con inserzioni di successo.
Utilizzando l’antibiotico puromicina o blasticidin con il kit, il team di Chesnut è riuscito ad aumentare il tasso di inserimento del gene dal 10-30% al 90% o più in alcune popolazioni di cellule. Alcuni geni particolarmente difficili sono passati da un tasso di inserimento di meno dell’1 per cento a più del 90 per cento. È importante testare più dosi di antibiotici sulla linea cellulare che si prevede di utilizzare per trovare la dose corretta, dice Chesnut: si desidera uccidere le cellule senza inserzioni, ma non le cellule con inserzioni di successo.
Prova: I marcatori selezionabili funzionano meglio quando il gene di interesse è altamente espresso, dice Chesnut. “Se non lo è, si può ancora ottenere la selezione, ma si potrebbe non ottenere abbastanza espressione del tag della proteina fluorescente per essere in grado di rilevarlo”. Inoltre, si applicano le limitazioni generali di CRISPR-Cas9. “Ci sono regioni del genoma che non tagliano molto bene con CRISPR, e non siamo ancora sicuri del perché”, aggiunge. E alcuni tipi di cellule non accettano facilmente DNA estraneo, RNA o complessi RNA-proteici – i tre metodi di consegna di CRISPR-Cas9.
Per avere più fortuna nell’inserire marcatori selezionabili, assicuratevi che ci sia una cosiddetta sequenza PAM, un breve tag nel DNA bersaglio che CRISPR-Cas9 deve riconoscere prima di tagliare, entro 10 coppie di basi del sito di inserimento del gene desiderato, dice Chesnut. Più lontano dal sito di taglio e l’efficienza di inserimento può essere troppo bassa per essere funzionale. Senza un sito PAM, si può provare TALENs o nucleasi a dito di zinco, anche se queste vecchie tecniche di editing genico sono più complicate di CRISPR.
Inibizione temporizzata
Ricercatore: Jacob Corn, biologo del genoma, Istituto Federale Svizzero di Tecnologia, Zurigo
Progetto: I ricercatori non capiscono perché la via NHEJ supera di gran lunga la via HDR nelle cellule dei mammiferi. “I lieviti fanno HDR come un matto”, dice Corn. Nel tentativo di accelerare questo processo di riparazione del DNA nelle cellule umane e migliorare il controllo dei geni knock-in, lui e il suo team stanno cercando di individuare come HDR è regolato. Hanno esaminato le cellule umane per i geni il cui knockdown ha portato ad un aumento di HDR nella cellula, e poi hanno cercato inibitori di piccole molecole di questi geni. Uno dei geni che è saltato fuori codifica per CDC7, una chinasi che regola la transizione del ciclo cellulare alla fase S; il suo inibitore, XL413, ha aumentato l’efficienza del gene knock-in di due o tre volte (BioRXiv, DOI: 10.1101/500462, 2018). Questo perché HDR si verifica solo in alcune parti del ciclo cellulare, compresa la fase S, dice Corn. Se si aggiunge l’inibitore XL413 nello stesso momento in cui si usa CRISPR-Cas9 per modificare il gene bersaglio, le cellule si accumulano nella fase immediatamente precedente alla fase S. Poi si rimuove XL413, e tutte le cellule vanno in fase S e aumentano l’efficienza knock-in.

Corn ha usato questa tecnica in molte linee cellulari umane immortalizzate e in cellule T umane. Può bussare a brevi tratti di DNA, come gli SNPs, così come a grandi geni. Non c’è motivo per cui non dovrebbe funzionare nei topi, dice, anche se non l’ha testato.
Prova: “Il tempismo è assolutamente fondamentale”, dice Corn. Il Cas9 deve tagliare il DNA nello stesso momento in cui viene aggiunto l’XL413. Se si inibisce prima e poi si rilascia durante l’editing con CRISPR-Cas9, l’efficienza della ricombinazione omologa si riduce di tre volte invece di aumentare, perché le cellule vengono rilasciate nella fase sbagliata del ciclo cellulare.
E come con qualsiasi sforzo HDR, Corn dice, esegui sempre un controllo senza nucleasi per assicurarti di non amplificare accidentalmente il DNA contaminante che galleggia nel tuo laboratorio. Dopo aver introdotto il knock-in, “sequenza, sequenza, sequenza, sequenza”, dice. Usare solo un sistema di segnalazione come una proteina fluorescente per dimostrare il successo dell’inserimento del gene può ritorcersi contro. Il sequenziamento verifica che le inserzioni sono state fatte nel sito corretto.
Giocando il gioco lungo
Ricercatore: Channabasavaiah Gurumurthy, direttore del centro di ingegneria del genoma del topo, Università del Nebraska Medical Center
Progetto: Alcuni anni fa, riflettendo sulla difficoltà di bussare ai geni cercando di farlo negli zigoti di topo, Gurumurthy e i suoi colleghi hanno avuto una rivelazione.
I ricercatori stavano inserendo con successo DNA corto e a singolo filamento, quindi perché non provare a fare un knock-in inserendo DNA lungo e a singolo filamento? In effetti, l’approccio, che Gurumurthy chiama Easi-CRISPR (aggiunte efficienti con inserti di ssDNA -CRISPR), aumenta l’efficienza di 2,5 volte, e usando il DNA a filo singolo taglia il tasso di inserzioni fuori bersaglio 100 volte nella cultura cellulare (Nat Protoc 13:195-215, 2018; Nature 559:405-09, 2018). “È abbastanza enorme”, dice. Nel laboratorio di Gurumurthy, Easi-CRISPR ha generato una linea di topo knock-in per 9 su 10 geni che hanno provato. Un collaboratore l’ha anche usata in cellule T umane per creare cellule CAR-T, cellule immunitarie specifiche del paziente per combattere il cancro.
Prova: Easi-CRISPR è tutt’altro che infallibile, avverte Gurumurthy. A volte la tecnica inserisce solo una parte del gene. Inoltre, aggiunge, può rimescolare i bracci di omologia, le brevi sequenze su entrambi i lati del gene che lo portano al suo obiettivo corretto nel genoma. E alcuni loci sono inspiegabilmente più difficili da inserire di altri.
Pochi fornitori commerciali progettano e sintetizzano DNA lungo e a singolo filamento. È possibile fare il proprio, ma la stabilità del DNA a singolo filamento varia; sequenze meno stabili avranno rendimenti inferiori, quindi potrebbe essere necessario sintetizzare più di loro, dice Gurumurthy.
I ricercatori incapaci di inserire CRISPR in embrioni di topo monocellulare possono pagare una struttura centrale per fare i topi con la loro sequenza di DNA, dice Gurumurthy. Le strutture di base come la sua carica da $5,000-$15,000 per generare una o due coppie di riproduzione; le strutture commerciali fanno pagare $20,000-$50,000, dice.
Knock-in By Numbers
Ricercatore: Greg Findlay, candidato MD/PhD nel laboratorio di Jay Shendure, Università di Washington
Progetto: Findlay e i suoi colleghi miravano a migliorare il modo in cui i medici interpretano le mutazioni nel gene del cancro al seno e alle ovaie BRCA1. Quel gene ha migliaia di varianti, ma i ricercatori non sanno come la maggior parte di loro influenzano la sua funzione. Per studiare l’impatto di queste varianti, hanno usato una tecnica knock-in che hanno sviluppato chiamata editing del genoma di saturazione (Nature, 562:217-22, 2018).
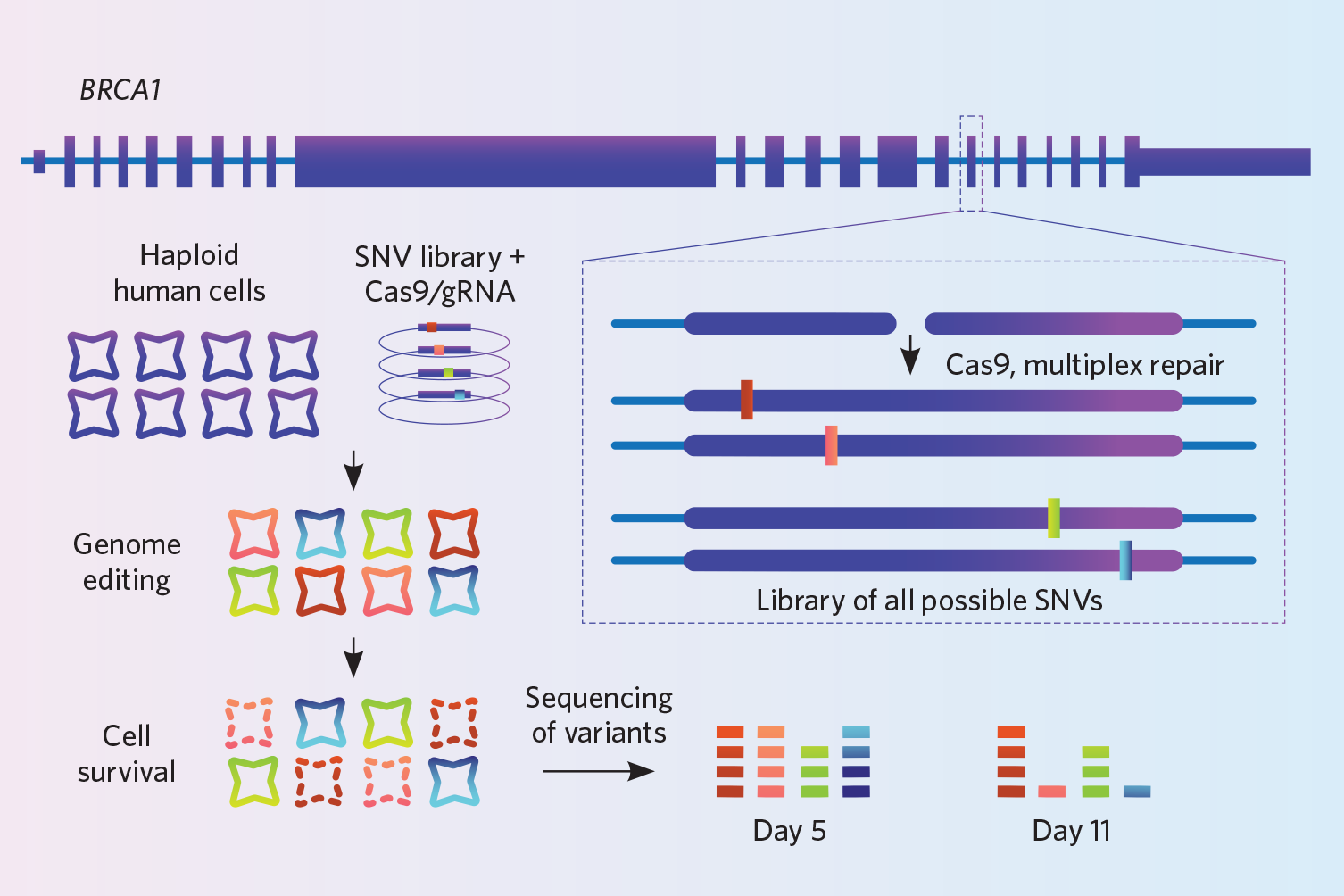
In una linea cellulare umana aploide immortalizzata, hanno usato CRISPR-Cas9 per battere in vitro 4.000 piccole varianti in milioni di cellule contemporaneamente. Il genoma viene tagliato nello stesso punto in ogni cellula, ma il genoma di ogni cellula riceve una variante diversa. Per promuovere l’HDR, hanno anche eliminato il gene ligase4, disabilitando la via di riparazione NHEJ – un passo che ha prodotto un guadagno di tre volte in efficienza, dice Findlay. Infine, poiché tutti i knock-in delle cellule sono diversi, hanno sequenziato le cellule in profondità, coprendo la stessa regione genomica milioni di volte, per essere sicuri di aver effettivamente bussato alle 4.000 varianti che volevano studiare. Hanno sequenziato in due momenti, e hanno dedotto che i knock-in che non sono venuti fuori nel sequenziamento al secondo momento erano quelli che hanno interferito con la funzione del gene, perché le cellule che li portano devono essere morti: Il team di Findlay ha fatto produrre gli oligo DNA per le 4.000 varianti su un microarray. È possibile acquistare array da 6.000 a 250.000 oligo, quindi considerare di ottenere più bang per il vostro dollaro combinando più esperimenti sullo stesso array, dice Findlay. Il loro laboratorio paga circa $5.000 per 100.000 oligos.
Questa strategia viene con limitazioni: finora è stato utilizzato solo per battere in varianti di singolo nucleotide, e tutte le modifiche devono essere nello stesso gene. Il metodo funziona meglio quando si modifica una regione abbastanza stretta di DNA, circa 110-120 paia di basi, perché gli oligo DNA più lunghi avrebbero troppi errori, dice Findlay. È anche importante sequenziare molto profondamente per assicurarsi che si tenga conto del numero completo di varianti che si intende battere in.
.