- A Clinicopathologic Review
- INTRODUZIONE
- DISTROFIE CORNEALI EPITELIALI-STROMALI
- DISTROFIA CORNEALE DI REIS-BUCKLERS
- DISTROFIA CORNEALE A LATTICE
- DISTRROFIA CORNEALE GRANULARE, TIPO I
- DISTROFIA CORNEALE GRANULARE, TIPO II
- DISTROFIE CORNEALI STROMALI
- DISTROFIA CORNEALE MACULARE
- DISTROFIA CORNEALE DI SCHNYDER (SCD)
- Tabella 4: Mnemonico per ricordare le distrofie stromali corneali
- OVERVIEW: DISTROFIE CORNEALI STROMALI
- EPIDEMIOLOGIA
- SIGNS
- SINTOMI
- TREATTAZIONE
A Clinicopathologic Review
Emily S. Birkholz, MD, Nasreen A. Syed, MD, and Michael D. Wagoner, MD, PhD
August 17, 2009
Major Revision: Chaunhi Van, MD e Nasreen Syed, MD
Agosto 20, 2015
INTRODUZIONE
Le distrofie epiteliali-stromali e stromali della cornea sono un gruppo di disturbi ereditari della cornea che sono causati dal progressivo accumulo di depositi negli strati della cornea. Questi depositi non sono causati da infiammazioni, infezioni o traumi, ma da mutazioni genetiche che portano alla trascrizione di proteine aberranti con conseguente accumulo di materiale insolubile nella cornea. I disturbi possono influire o meno sulla visione e possono essere simmetrici o meno (1). Il sistema di classificazione dell’International Committee for Classification of Corneal Dystrophies (IC3D) del 2015 ha suddiviso le distrofie corneali in 4 categorie: distrofie epiteliali e subepiteliali, distrofie epitelio-stromali, distrofie stromali e distrofie endoteliali. La maggior parte delle distrofie precedentemente considerate stromali sono ora classificate come distrofie epitelio-stromali o distrofie stromali. Le tabelle 1 e 2 elencano le distrofie epitelio-stromali e le distrofie stromali (2). La vecchia classificazione delle distrofie stromali corneali è elencata nella tabella 3.
- Distrofia corneale di Reis-Bucklers
- Distrofia corneale di Thiel-Behnke
- Distrofia corneale di Lattice, tipo 1 e varianti
- Distrofia corneale granulare, tipo 1
- Distrofia corneale granulare, tipo 2
- Distrofia corneale maculare
- Distrofia corneale di Schnyder
- Distrofia corneale stromale congenita
- Distrofia corneale amorfa posteriore
- Distrofia torbida centrale di Francois
- Pre-Distrofia corneale di Descemet
Tabella 3. Vecchia classificazione delle distrofie corneali stromali
- Distrofia corneale a reticolo
- Distrofia corneale granulare
- Distrofia corneale di Avellino
- Distrofia corneale maculare
- Distrofia gelatinosa a goccia
- Distrofia corneale a gocciacome la distrofia
- Distrofia corneale di Schnyder
- Distrofia di Francois-Neetans Fleck
- Distrofia stromale ereditaria congenita
DISTROFIE CORNEALI EPITELIALI-STROMALI
Le distrofie epiteliali-stromali sono causate da mutazioni nel gene TGFβI (transforming growth factor beta-induced), conosciuto anche come il gene BIGH3. TGFβI si trova sul cromosoma 5q31 e codifica per la cheratoepitelina, una proteina secreta dall’epitelio corneale. Questa proteina agisce come una proteina di adesione ed è presente nello stroma normale. Essendo una piccola proteina delle dimensioni dell’albumina, ha la capacità di diffondersi attraverso lo stroma corneale. Quando si verifica una mutazione nel gene TGFβI, la struttura della cheratoepitelina è anormale e si verifica un accumulo della proteina insolubile o dei suoi frammenti proteolitici nella cornea (1, 3). È interessante notare che la mutazione del gene TGFβI è stata scoperta in parte all’Università dell’Iowa. Un gruppo di ricercatori e clinici tra cui Edwin M. Stone, Robert Folberg e Jay H. Krachmer ha mappato la distrofia granulare di tipo I, la distrofia granulare di tipo II e la distrofia reticolare sul cromosoma 5q nel 1994 (4). Ad oggi, 63 diverse mutazioni sono state identificate nel gene TGFβI. Non sono stati identificati trattamenti efficaci per prevenire o attenuare la deposizione della cheratoepitelina. Le distrofie hanno tipicamente un’eredità autosomica dominante e coinvolgono lo strato di Bowman e lo stroma (3).
DISTROFIA CORNEALE DI REIS-BUCKLERS
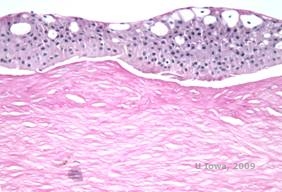
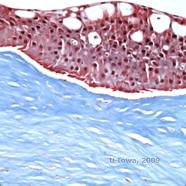
Reis-Bücklers, precedentemente conosciuta come distrofia corneale granulare di tipo III o distrofia corneale di Bowman di tipo I, si presenta tipicamente con cornee normali alla nascita ma sviluppa erosioni ricorrenti dolorose, opacizzazione e perdita progressiva della vista entro la prima decade di vita (1). Le opacità irregolari, grigio-bianche, di tipo geografico, si trovano nello strato di Bowman e nello stroma anteriore. Nelle fasi più avanzate della malattia, le opacità possono estendersi al limbus e allo stroma più profondo (2). L’istopatologia rivela depositi stromali anteriori e subepiteliali di materiale simile alla ialina che interrompono e spesso sostituiscono lo strato di Bowman (vedi Figura 1A e 1B). I depositi si colorano di rosso con la colorazione tricromica di Masson (2). Il materiale ialino è costituito ultrastrutturalmente da corpi simili a bastoncini, il che aiuta a distinguerlo dalla distrofia corneale di Thiel-Behnke (1, 2).
| A: H&E di Reis Bückler che mostra distruzione dello strato di Bowman ed epitelio irregolare | B. Colorazione tricromica di Masson che dimostra la colorazione epiteliale |
 |
 |
DISTROFIA CORNEALE A LATTICE
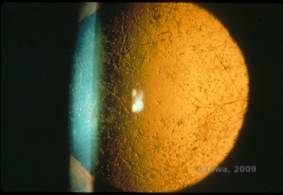
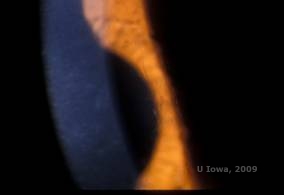
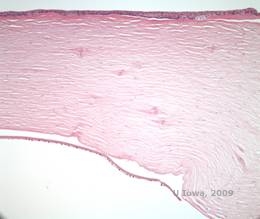
La distrofia corneale a reticolo (LCD) è la più comune delle distrofie epiteliali-stromali della cornea. È tipicamente una malattia autosomica dominante e bilaterale che si presenta verso la fine della prima decade di vita con sintomi di erosioni corneali ricorrenti e diminuzione della vista. È caratterizzata da linee reticolari che sono opacità rifrangenti lineari, orientate radialmente e ramificate, descritte come “simili al vetro”, situate nello stroma anteriore (vedi Figura 2A e 2B). Queste linee reticolari si trovano inizialmente nella cornea centrale superficiale. Con il progredire della malattia, si diffondono più profondamente e perifericamente nello stroma con risparmio del limbus (1, 2). Altri risultati dell’esame includono opacità simili a macchie, punti bianchi subepiteliali e foschia stromale “ground-glass”, che inizia centralmente e diventa più diffusa (2). Molti pazienti con LCD richiederanno un intervento chirurgico per il trattamento delle erosioni ricorrenti e della diminuzione della vista. Se la malattia è localizzata anteriormente nello stroma, i pazienti possono spesso essere trattati con successo con la cheratectomia fototerapeutica (PTK). Alcuni richiedono il trapianto di cornea. Poiché la cheratoepitelina, la proteina prodotta dal gene TGFβI, è prodotta principalmente nell’epitelio corneale, la malattia tende a ripresentarsi negli innesti corneali (1).
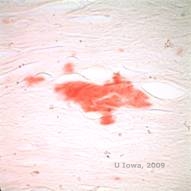
Nella LCD, i depositi amiloidi si accumulano tra la membrana di base epiteliale e lo strato di Bowman, nonché nello stroma, causando la distorsione dell’architettura lamellare. I depositi si colorano positivamente con l’immunoistochimica usando anticorpi contro la cheratoepitelina (2). I depositi appaiono come depositi rosa amorfi alla colorazione con ematossilina ed eosina (H&E) (vedi Figura 1C e 1D) e si colorano di rosso Congo dimostrando la classica birifrangenza verde mela alla polarizzazione incrociata (vedi Figura 2E e 2F) (1). Assenza o assottigliamento dello strato di Bowman, atrofia epiteliale e degenerazione epiteliale basale possono anche essere trovati all’istopatologia in LCD (2).
LCD tipo I è la forma classica di LCD causata da una mutazione nel gene TGFβI con conseguente deposito isolato di amiloide nella cornea. Sono state identificate quattro varianti di LCD: LCD tipo IIIA, tipo I/IIIA, tipo IV e amiloidosi polimorfica. Le varianti dell’LCD si presentano più tardi nella vita rispetto all’LCD classico. L’LCD tipo IIIA si presenta nella 5°-7° decade, di solito con erosioni epiteliali. Ha linee reticolari più spesse, descritte come “ropy-appearing”, che si estende al limbus. L’LCD di tipo I/IIIA ha linee reticolari sottili. L’LCD di tipo IV si presenta nella 7a-9a decade con piccole linee reticolari. I depositi di amiloide nell’LCD di tipo IV si trovano nello stroma profondo e raramente si verificano erosioni epiteliali. Le linee reticolari sono assenti nell’amiloidosi di tipo polimorfico e raramente si verificano erosioni epiteliali (2).
L’LCD tipo II è una sindrome da amiloidosi sistemica nota come sindrome di Meretoja che colpisce la pelle, i nervi cranici e la cornea. Si presenta nella prima età adulta con neuropatie periferiche, neuropatie craniche, facies da segugio, pelle secca, blefarochalasi, labbra sporgenti e linee reticolari corneali. Questo tipo è stato collegato al gene della gelsolina sul cromosoma 9, che codifica per una proteina precursore dell’amiloide che funziona per rimuovere l’actina dai siti di lesione e infiammazione (1). Il nome è un termine improprio e non è considerato una variante della distrofia corneale a reticolo (2).
| A: Occhio sinistro su retroilluminazione che dimostra depositi stromali anteriori nella distrofia corneale a reticolo | B: Occhio sinistro con maggiore potenza che mostra depositi stromali anteriori lineari. |
 |
 |
| C: Macchia H&E di cornea con reticolo. Notare i depositi amorfi rosa nello stroma | D: Una vista più ravvicinata dei depositi amorfi rosa |
 |
 |
| E: Macchia rosso Congo, che evidenzia l’amiloide | F: Birifrangenza verde mela dell’amiloide con polarizzazione incrociata. |
 |
 |
DISTRROFIA CORNEALE GRANULARE, TIPO I
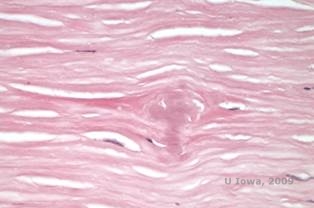
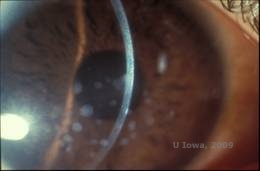
Distrofia corneale granulare, tipo I (GCD1) è una malattia bilaterale, autosomica dominante associata ad una mutazione nel gene TGFβI che porta alla deposizione di un materiale ialino nello stroma corneale. Si presenta tipicamente all’inizio della prima decade di vita con opacità grigio-bianche, “simili a briciole” nello stroma anteriore e medio, estendendosi nello stroma posteriore nella malattia avanzata (1, 2). Queste opacità sono depositi discreti situati centralmente, con cornea chiara situata nella periferia e cornea chiara tra i depositi (vedi Figura 3A e 3B). La malattia è tipicamente asintomatica all’inizio, ma con il tempo le opacità possono coalizzarsi e portare a una diminuzione della vista. Erosioni corneali ricorrenti possono verificarsi nel GCD, ma con un’incidenza inferiore rispetto al LCD (1, 5). I pazienti possono anche sperimentare abbagliamento e fotofobia (2). Il trattamento all’inizio del processo della malattia è spesso solo l’osservazione. Tuttavia, con il progredire della malattia, la PTK e il trapianto di cornea possono essere necessari per migliorare la visione e i sintomi di erosione. Come l’LCD, la malattia può ripresentarsi negli innesti corneali.
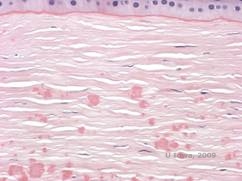
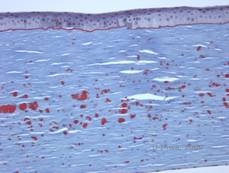
Istopatologicamente le opacità sono depositi eosinofili spesso descritti come “rock candy like” nello stroma anteriore fatto di un materiale ialino. Con il tempo, i depositi progrediscono nello stroma corneale più profondo. Il materiale ialino si macchia di rosso vivo con la colorazione tricromica di Masson (vedi Figura 3C e 3D).
| A:Foto alla lampada a fessura della distrofia corneale granulare, tipo I | B: Notare i depositi stromali “crumb-like” con stroma chiaro intermedio. |
 |
 |
| C: Colorazione H& E della cornea che mostra depositi ialini eosinofili “rock candy like” nello stroma | D: Materiale ialino si macchia di rosso vivo con Masson-Trichrome |
 |
 |
DISTROFIA CORNEALE GRANULARE, TIPO II
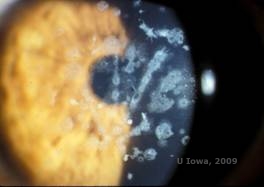
Distrofia corneale granulare, tipo II (GCD2), precedentemente conosciuta come Avellino o distrofia corneale combinata granulo-lattice, è una malattia autosomica dominante legata ad una mutazione nel gene TGFβI che porta ad un deposito sia ialino che amiloide nello stroma corneale. Tipicamente, i pazienti si presentano nella seconda decade di vita con piccole macchie grigio-bianche nello stroma superficiale. Le opacità possono anche apparire di forma spinosa, anulare o stellata. In retroilluminazione, sono parzialmente traslucide. Più tardi nel processo della malattia, possono anche sviluppare linee reticolari (vedi Figura 4A e 4B). Queste linee non si incrociano e appaiono più bianche e meno rifrangenti delle linee reticolari. I sintomi del GCD2 sono il dolore con erosioni epiteliali e la diminuzione della vista (2).
Istopatologicamente, la cornea avrà depositi stromali che si colorano di rosso con il tricromo di Masson, indicando la presenza di ialino (Vedi Figura 4C). Inoltre, la colorazione con il rosso Congo dimostrerà una birifrangenza verde mela sulla polarizzazione incrociata che indica la presenza di amiloide (Vedi Figura 4D). Si pensava che la malattia avesse avuto origine da una famiglia di Avellino, in Italia. Tuttavia, il GCD di tipo II è stato ora riportato anche in pazienti di molti altri paesi (2,5), con la più alta prevalenza nell’Asia orientale.
| A: Distrofia di Avellino che mostra depositi di tipo reticolare e granulare nello stroma corneale | B. Macchia tricromica di Masson che dimostra depositi ialini stromali anteriori |
 |
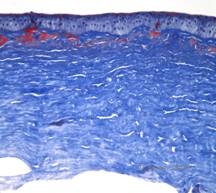 |
| C: Macchia rosso Congo che mostra depositi amorfi amiloidi rosa nello stesso campione corneale. | D: La polarizzazione incrociata rivela una birifrangenza verde mela che indica l’amiloide. |
 |
 |
DISTROFIE CORNEALI STROMALI
DISTROFIA CORNEALE MACULARE
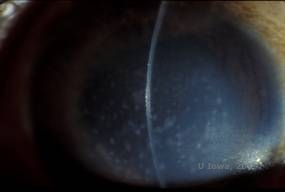
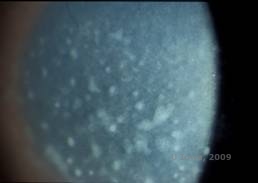
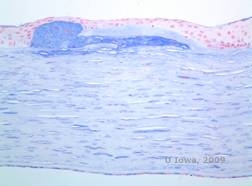
La distrofia corneale maculare (MCD) è una malattia autosomica recessiva causata da una mutazione nel gene della carboidrato sulfotransferasi 6 (CHST6) sul cromosoma 16 che porta ad un difetto nella sintesi del solfato di cheratano, il principale glicosaminoglicano della cornea. È meno comune del LCD o del GCD, ma tende ad avere un impatto più grave sulla vista. Sebbene la MCD sia meno comune in tutto il mondo rispetto alla LCD o alla GCD, è la più comune delle distrofie stromali corneali in luoghi come l’Islanda e l’Arabia Saudita (2,6). Le lesioni stromali anteriori di colore grigio-bianco, simili al GCD1, appaiono nella cornea nella prima decade di vita. A differenza del GCD1, tuttavia, c’è una foschia stromale tra i depositi e l’intera cornea dal limbus al limbus è spesso coinvolta (vedi Figura 5A e 5B). La cornea è sottile e con il progredire del disturbo la membrana di Descemet diventa grigia e sviluppa guttae. Possono verificarsi erosioni epiteliali, ma meno nella MCD che nella LCD. I pazienti sviluppano tipicamente una grave perdita visiva entro la seconda o terza decade di vita a causa della diffusa foschia corneale. La PTK può essere eseguita in alcuni casi precoci di MCD. Tuttavia, questa condizione non è in genere suscettibile di PTK come la distrofia reticolare o granulare e spesso richiede il trapianto di cornea per il trattamento (7). La recidiva negli innesti è meno comune nella MCD che nella distrofia granulare o reticolare (1,2,5,6,8).
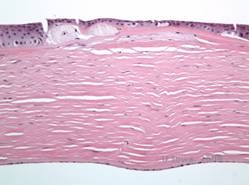
I depositi stromali nella MCD sono composti da mucopolisaccaridi che si accumulano nel reticolo endoplasmatico dei cheratociti dello stroma corneale, extracellularmente tra le lamelle stromali e nell’epitelio, nella membrana Descemet e nell’endotelio. Questi depositi si colorano di blu Alcian (vedi figura 5C e 5D) (1). Ci sono rotture nello strato di Bowman e nelle guttae con ispessimento della membrana di Descemet (2).
Tre sottotipi di MCD sono stati descritti in base alla presenza o assenza di cheratano solfato immunoreattivo in vari tessuti. Il tipo I non ha cheratano solfato immunoreattivo nello stroma corneale, nei cheratociti, nei sieri o nella cartilagine, ed è la variante più comune di MCD in tutto il mondo. Il tipo IA manca di cheratano solfato nello stroma, nei sieri e nella cartilagine, ma ha livelli rilevabili nei cheratociti. Il tipo II ha cheratano solfato presente a livelli molto ridotti nello stroma, nei cheratociti, nei sieri e nella cartilagine (6).
| A: Foto con lampada a fessura della distrofia corneale maculare. | B: Notare la foschia tra i depositi stromali corneali |
 |
 |
| C: H&E di cornea con distrofia maculare. Notare i depositi stromali anteriori e la rottura dello strato di Bowman | D: Depositi di mucopolisaccaridi all’interno dei cheratociti evidenziati con la colorazione Alcian Blue |
 |
 |
DISTROFIA CORNEALE DI SCHNYDER (SCD)
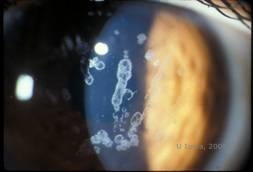
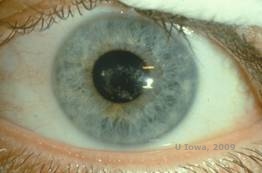
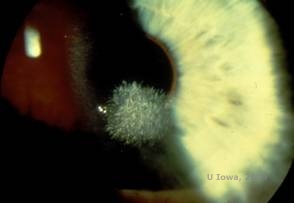
Distrofia corneale di Schnyder (SCD), precedentemente conosciuta come distrofia corneale cristallina di Schnyder, è una distrofia corneale stromale bilaterale autosomica dominante, legata ad una mutazione genetica nel gene UbiA preniltransferasi contenente il dominio 1 (UBIAD1) sul cromosoma 1. Il conseguente difetto metabolico dei cheratociti corneali porta alla deposizione di colesterolo cristallino nello stroma. Tuttavia, la presenza di cristalli non è assolutamente necessaria per la diagnosi di SCD. Infatti, solo il 54% dei pazienti con SCD ha cristalli corneali. Tipicamente, i pazienti si presentano nella seconda o terza decade con un’opacità corneale centrale a forma di anello con o senza cristalli sottoepiteliali a forma di virgola (vedi Figura 6A e 6B). Poi, l’arcus lipoides appare tra i 23 e i 38 anni. Dopo i 38 anni, il progressivo intorbidimento corneale si traduce in una foschia panstromale che raggiunge la media periferia. La maggior parte dei pazienti oltre i 50 anni di età ha una perdita della visione fotopica, abbagliamento e diminuzione della sensazione corneale, e quindi, può richiedere un trattamento chirurgico tra cui il trapianto di cornea o la PTK. Possono verificarsi recidive nell’innesto. La malattia è stata collegata con ipercolesterolemia, iperlipidemia e genu valgum in alcuni pazienti (2,5,9,10).
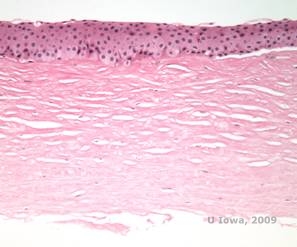
Istopatologicamente, cristalli di colesterolo birifrangenti composti da fosfolipidi e colesterolo si depositano nelle cellule epiteliali basali, nei cheratociti, nello strato di Bowman e tra le lamelle stromali. I lipidi si dissolvono nel normale trattamento istologico, quindi devono essere ottenute sezioni congelate attraverso la cornea per dimostrare la presenza di lipidi con macchie Oil-Red-O o Sudan black.
| A: Foto con lampada a fessura della distrofia corneale di Schnyder. | B. Depositi cristallini localizzati centralmente |
 |
 |
| C: H&E di cornea con SCCD | D. La macchia Oil Red O evidenzia i cristalli di colesterolo che appaiono rossi. |
 |
 |
La tabella 4 fornisce un mnemonico comune per memorizzare alcune delle distrofie corneali che colpiscono lo stroma, la composizione del loro deposito e il metodo di colorazione di questi depositi è elencato.
Tabella 4: Mnemonico per ricordare le distrofie stromali corneali
- Distrofia Marilyn-Maculare
- Monroe-Mucopolisaccaride
- Sempre colorazione Blu Alcian
- Gets-Granular Distrofia
- Her-Hyaline
- Macchia tricromica Man in-Masson
- Distrofia Los-Lattice
- Angeles-Amyloid
- California-Congo Red
OVERVIEW: DISTROFIE CORNEALI STROMALI
EPIDEMIOLOGIA
|
SIGNS
|
SINTOMI
|
TREATTAZIONE
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Patologia congiuntivale e corneale. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3a edizione. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D classification of corneal dystrophies–edition 2. Cornea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. Aspetti clinici e genetici delle distrofie corneali associate al TGFBI. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Tre distrofie corneali autosomiche dominanti mappa al cromosoma 5q. Natura genet. 1994; 6: 47-51.
- Bron AJ. Genetica delle distrofie corneali: Cosa abbiamo imparato negli ultimi venticinque anni. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Cheratoplastica penetrante per la distrofia corneale maculare. Ophthalmology 2005;112: 220-224.
- Badr IA, Wagoner MD. Cheratectomia fototerapeutica per la distrofia corneale maculare. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetica delle distrofie corneali anteriori e stromali. Seminari in oftalmologia, 2008; 23: 1,9-17.
- Weiss JS. Distrofia corneale di Schnyder. Curr Opin Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. Il gene per la distrofia corneale cristallina di Schnyder mappa al cromosoma umano 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Formato di citazione suggerito: Van C, Syed NA. Distrofie corneali epiteliali-stromali e stromali: Una revisione clinicopatologica. Revisione di ; EyeRounds.org. 20 agosto 2015. Disponibile da: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm