- Klinikkapatologinen katsaus
- TIIVISTELMÄ
- EPITELIAL-STROMAL CORNEAL DYSTROPHIES
- REIS-BUCKLERSIN SORNAALIDYSTROFIA
- LATTICE CORNEAL DYSTROPHY
- GRANULAARINEN SYVÄKALVODYSTROFIA, TYYPPI I
- Granulaarinen sarveiskalvon dystrofia, tyyppi II
- STROMAALINEN SYVÄKALVODYSTROFIA
- MAKULAARINEN SYVÄKALVODYSTROFIA
- SCHNYDERIN SYRVÄKALVODYSTROFIA (SCD)
- Taulukko 4: Muistilista sarveiskalvon stroomadystrofioiden muistamista varten
- OVERVIEW: CORNEAL STROMAL DYSTROPHIES
- EPIDEMIOLOGIA
- MERKITSEVÄT
- SYMPTOMIT
- HOITO
Klinikkapatologinen katsaus
Emily S. Birkholz, MD, Nasreen A. Syed, MD, ja Michael D. Wagoner, MD, PhD
17. elokuu 2009
Pääasiallinen tarkistus: Chaunhi Van, MD ja Nasreen Syed, MD
20. elokuuta 2015
TIIVISTELMÄ
Sarveiskalvon epiteeli-stroomaaliset ja stroomaaliset dystrofiat ovat ryhmä sarveiskalvon perinnöllisiä sairauksia, jotka johtuvat kerrostumien etenevästä kertymisestä sarveiskalvon kerroksiin. Nämä kerrostumat eivät johdu tulehduksesta, infektiosta tai traumasta, vaan geneettisistä mutaatioista, jotka johtavat poikkeavien proteiinien transkriptioon, mikä johtaa liukenemattoman materiaalin kertymiseen sarveiskalvoon. Häiriöt voivat vaikuttaa tai olla vaikuttamatta näkökykyyn, ja ne voivat olla symmetrisiä tai epäsymmetrisiä (1). Sarveiskalvon dystrofioiden kansainvälisen luokittelukomitean (International Committee for Classification of Corneal Dystrophies, IC3D) vuonna 2015 laatimassa luokitusjärjestelmässä sarveiskalvon dystrofiat on jaettu neljään luokkaan: epiteliaaliset ja subepiteliaaliset dystrofiat, epiteliaalis-stroomaaliset dystrofiat, stroomaaliset dystrofiat ja endoteelialiset dystrofiat. Useimmat dystrofiat, joita aiemmin pidettiin stromaalisina, luokitellaan nyt joko epiteelis-stromaalisiksi dystrofioiksi tai stromaalisiksi dystrofioiksi. Taulukoissa 1 ja 2 luetellaan epiteeli-stroomaaliset dystrofiat ja stroomaaliset dystrofiat (2). Sarveiskalvon stroomadystrofioiden vanha luokitus on lueteltu taulukossa 3.
- Reis-Bucklersin sarveiskalvon dystrofia
- Thiel-Behnken sarveiskalvon dystrofia
- Lattice-sarveiskalvon dystrofia, tyyppi 1 ja muunnokset
- Granulaarinen sarveiskalvon dystrofia, tyyppi 1
- Granulaarinen sarveiskalvon dystrofia, tyyppi 2
Taulukko 3. Sarveiskalvon stroomadystrofioiden vanha luokitus
- Lattice-sarveiskalvon dystrofia
- Granular-sarveiskalvon dystrofia
- Avellinon sarveiskalvon dystrofia
- Makulaarinen sarveiskalvon dystrofia
- Gelatiinipisara-dystrofia
- Schnyderin sarveiskalvon dystrofia
- Francois-Neetans Fleck dystrophy
- Congenital hereditary stromal dystrophy
EPITELIAL-STROMAL CORNEAL DYSTROPHIES
Epiteliaalis-stroomaaliset dystrofiat johtuvat transformoivan kasvutekijän beeta-indusoiman (TGFβI) geenin mutaatioista, joka tunnetaan myös nimellä BIGH3-geeni. TGFβI sijaitsee kromosomissa 5q31 ja koodaa keratoepiteliiniä, sarveiskalvon epiteelin erittämää proteiinia. Tämä proteiini toimii adheesioproteiinina, ja sitä esiintyy normaalissa stroomassa. Koska se on pieni, suunnilleen albumiinin kokoinen proteiini, se pystyy diffundoitumaan sarveiskalvon strooman läpi. Kun TGFβI-geenissä tapahtuu mutaatio, keratoepiteliinin rakenne on epänormaali ja sarveiskalvoon kertyy liukenematonta proteiinia tai sen proteolyyttisiä fragmentteja (1, 3). Mielenkiintoista on, että TGFβI-geenin mutaatio löydettiin osittain Iowan yliopistossa. Ryhmä tutkijoita ja lääkäreitä, joihin kuuluivat Edwin M. Stone, Robert Folberg ja Jay H. Krachmer, kartoitti granulaarisen tyypin I, granulaarisen tyypin II ja lattice-dystrofian kromosomille 5q vuonna 1994 (4). Tähän mennessä TGFβI-geenistä on tunnistettu 63 erilaista mutaatiota. Tehokkaita hoitoja keratoepiteliinin kerrostumisen estämiseksi tai lieventämiseksi ei ole tunnistettu. Dystrofiat ovat tyypillisesti autosomaalisesti dominantisti periytyviä, ja ne koskevat Bowmanin kerrosta ja stroomaa (3).
REIS-BUCKLERSIN SORNAALIDYSTROFIA
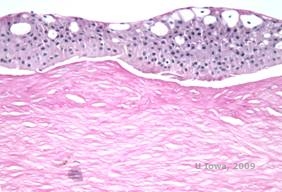
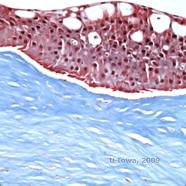
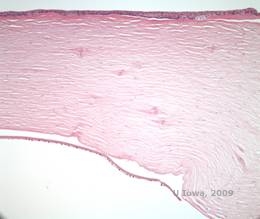
Reis-Bücklers, aiemmin tunnettu nimellä Granulaarinen sarveiskalvon dystrofia tyyppi III tai Bowmanin tyypin I sarveiskalvon dystrofia, esiintyy tyypillisesti normaaleina sarveiskalvoina syntyessään, mutta niille kehittyy kivuliaita, toistuvia eroosioita, sameutta ja etenevää näön heikkenemistä ensimmäisellä vuosikymmenellä elämästä (1). Bowmanin kerroksessa ja anteriorisessa stroomassa on epäsäännöllisiä, harmaanvalkoisia, maantieteellisen kaltaisia samentumia. Taudin edistyneemmissä vaiheissa opasiteet voivat ulottua limbukseen ja syvemmälle stroomaan (2). Histopatologiassa havaitaan anteriorisen strooman ja subepiteelin hyaliinin kaltaisen materiaalin kerrostumia, jotka häiritsevät Bowmanin kerrosta ja usein korvaavat sen (ks. kuvat 1A ja 1B). Kerrostumat värjäytyvät punaisiksi Massonin trikromivärjäyksellä (2). Hyaliinin kaltainen materiaali koostuu ultrastruktuurisesti sauvamaisista kappaleista, mikä auttaa erottamaan sen Thiel-Behnken sarveiskalvon dystrofiasta (1, 2).
| A: H&E Reis Bücklerin sarveiskalvon dystrofia, jossa näkyy Bowmanin kerroksen tuhoutuminen ja epäsäännöllinen epiteeli | B. Masson Trichrome -värjäys, joka osoittaa epiteelin värjäytymistä |
 |
 |
LATTICE CORNEAL DYSTROPHY
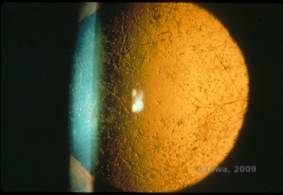
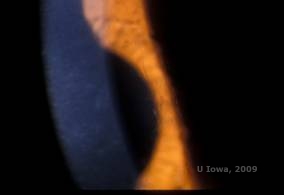
Ristikkosarveiskalvon dystrofia (LCD, Lattice Corneal Dystrophy) on tavallisin sarveiskalvojen epiteelistroomaalisista stromaalisista dystrofeista. Se on tyypillisesti autosomaalisesti dominoiva, molemminpuolinen sairaus, joka tyypillisesti ilmaantuu ensimmäisen elinvuosikymmenen loppupuolella ja jonka oireina ovat toistuvat sarveiskalvon eroosiot ja näön heikkeneminen. Sille on ominaista ristikkoviivat, jotka ovat lineaarisia, säteittäisesti suuntautuneita, haarautuvia, taittuvia, ”lasimaisiksi” kuvattuja samentumia, jotka sijaitsevat sarveiskalvon etuosan stroomassa (ks. kuvat 2A ja 2B). Näitä ristikkoviivoja esiintyy aluksi sarveiskalvon pinnallisessa keskiosassa. Taudin edetessä ne leviävät syvemmälle ja perifeerisesti stroomaan ja säästävät limbusta (1, 2). Muita tutkimuslöydöksiä ovat laikkumaiset samentumat, subepiteelialueen valkoiset pisteet ja ”ground-glass” -strooman sameus, joka alkaa keskeltä ja muuttuu hajanaisemmaksi (2). Monet LCD-potilaat tarvitsevat kirurgisen toimenpiteen toistuvien eroosioiden ja heikentyneen näön hoitamiseksi. Jos tauti sijaitsee anteriorisesti stroomassa, potilaita voidaan usein hoitaa menestyksekkäästi valoterapeuttisella keratektomialla (PTK). Jotkut tarvitsevat sarveiskalvonsiirtoa. Koska TGFβI-geenin tuottama proteiini keratoepiteliini tuotetaan enimmäkseen sarveiskalvon epiteelissä, taudilla on taipumus uusiutua sarveiskalvosiirroissa (1).
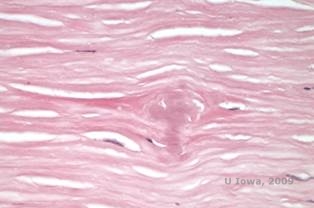
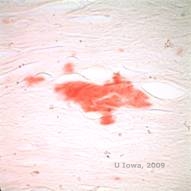
LCD:ssä amyloidikerrostumat kerääntyvät epiteelin tyvikalvon ja Bowmanin kerroksen väliin sekä sarveiskalvon stroomaan aiheuttaen lamelliarkkitehtuurin vääristymistä. Kerrostumat värjäytyvät positiivisesti immunohistokemiassa, jossa käytetään vasta-aineita keratoepiteliiniä vastaan (2). Kerrostumat näkyvät amorfisina vaaleanpunaisina kerrostumina hematoksyliini- ja eosiinivärjäyksessä (H&E) (ks. kuvat 1C ja 1D), ja ne värjäytyvät kongopunavärjäyksellä, joka osoittaa klassista omenanvihreää kaksoiskuviointia ristipolarisaatiossa (ks. kuvat 2E ja 2F) (1). LCD:n histopatologiassa voidaan havaita myös Bowmanin kerroksen puuttumista tai ohenemista, epiteelin atrofiaa ja basaaliepiteelin rappeutumista (2).
LCD tyyppi I on LCD:n klassinen muoto, joka johtuu TGFβI-geenin mutaatiosta, joka johtaa sarveiskalvon yksittäiseen amyloidikerrostumaan. Neljä LCD-muunnosta oli tunnistettu: LCD-tyyppi IIIA, tyyppi I/IIIA, tyyppi IV ja polymorfinen amyloidoosi. LCD-muunnokset ilmenevät myöhemmin elämässä kuin klassinen LCD. LCD-tyyppi IIIA ilmaantuu 5.-7. vuosikymmenellä, ja siihen liittyy yleensä epiteelin eroosioita. Siinä on paksumpia ristikkoviivoja, joita kuvataan ”köynnösmäisiksi” ja jotka ulottuvat limbukseen asti. LCD-tyypin I/IIIA:ssa on ohuita ristikkoviivoja. LCD-tyyppi IV esiintyy 7.-9. vuosikymmenellä, ja siinä on pieniä ristikkoviivoja. LCD-tyypin IV amyloidikerrostumia esiintyy syvällä stroomassa, ja epiteelin eroosiota esiintyy harvoin. Polymorfisessa amyloidoosityypissä ei ole ristikkoviivoja, ja epiteelin eroosioita esiintyy harvoin (2).
LCD-tyyppi II on systeeminen amyloidoosioireyhtymä, joka tunnetaan nimellä Meretojan oireyhtymä ja joka vaikuttaa ihoon, kallohermoihin ja sarveiskalvoon. Se ilmenee varhaisaikuisuudessa ja siihen liittyy perifeerisiä neuropatioita, kraniaalisia neuropatioita, koiran näköinen ilme, kuiva iho, blefarokalaasi, ulkonevat huulet ja sarveiskalvon ristikkoviivat. Tämä tyyppi on yhdistetty kromosomissa 9 sijaitsevaan gelsoliinigeeniin, joka koodaa amyloidin esiasteproteiinia, jonka tehtävänä on irrottaa aktiiniä vaurio- ja tulehduskohdista (1). Nimi on virheellinen, eikä sitä pidetä ristikkosarveiskalvon dystrofian muunnoksena (2).
| A: Vasen silmä retroilluminaatiossa, joka osoittaa lattice-sarveiskalvon dystrofiassa esiintyviä anteriorisia stroomakertymiä | B: Vasen silmä suuremmalla potenssilla, jossa näkyy lineaariset anterioriset stroomakertymät. |
 |
 |
| C: H&E-värjäys sarveiskalvosta, jossa on ristikko. Huomaa vaaleanpunaiset amorfiset kerrostumat stroomassa | D: Lähempi näkymä vaaleanpunaisista, amorfisista kerrostumista |
 |
 |
| E: Kongopunavärjäys, joka korostaa amyloidia | F: Omenanvihreää amyloidin kaksoiskuviointia ristipolarisaatiolla. |
 |
 |
GRANULAARINEN SYVÄKALVODYSTROFIA, TYYPPI I
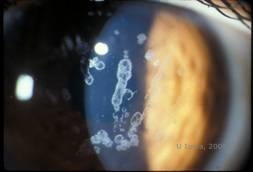
Granulaarinen sarveiskalvon dystrofia, tyyppi I (GCD1) on molemminpuolinen, autosomaalisesti dominoiva sairaus, joka liittyy TGFβI-geenin mutaatioon, joka johtaa hyaliinimaisen materiaalin kerrostumiseen sarveiskalvon stroomaan. Se ilmenee tyypillisesti varhain ensimmäisellä vuosikymmenellä harmaanvalkoisina, ”murumaisina” samentumina anteriorisessa ja keskimmäisessä stroomassa, jotka etenevässä taudissa laajenevat posterioriseen stroomaan (1, 2). Nämä samentumat ovat keskellä sijaitsevia erillisiä kerrostumia, joiden periferiassa on kirkas sarveiskalvo ja kerrostumien välissä kirkas sarveiskalvo (ks. kuvat 3A ja 3B). Tauti on tyypillisesti oireeton alkuvaiheessa, mutta ajan myötä opasiteet voivat sulautua yhteen ja johtaa näön heikkenemiseen. GCD:ssä voi esiintyä toistuvia sarveiskalvon eroosioita, mutta niiden esiintyvyys on pienempi kuin LCD:ssä (1, 5). Potilaat voivat myös kokea häikäisyä ja valonarkuutta (2). Hoito tautiprosessin alkuvaiheessa on usein pelkkää tarkkailua. Taudin edetessä voidaan kuitenkin tarvita PTK:ta ja sarveiskalvonsiirtoa näön ja eroosio-oireiden parantamiseksi. LCD:n tavoin tauti voi uusiutua sarveiskalvosiirteissä.
Histopatologisesti sameudet ovat eosinofiilisiä kerrostumia, joita kuvataan usein ”kivikaramellimaisiksi” anteriorisessa stroomassa, joka on valmistettu hyaliinin kaltaisesta materiaalista. Ajan myötä kerrostumat etenevät syvemmälle sarveiskalvon stroomaan. Hyaliinimateriaali värjäytyy kirkkaanpunaiseksi Masson-trikromivärjäyksellä (ks. kuvat 3C ja 3D).
| A: Viiltolamppukuva granulaarisesta sarveiskalvon dystrofiasta, tyyppi I | B: Huomaa ”murumaiset” stroomakertymät, joiden välissä on kirkas strooma. |
 |
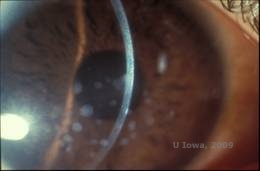 |
| C: Sarveiskalvon H& E-värjäys, jossa näkyy eosinofiiliset ”kivikarkkimaista karkkia muistuttavat” hyaliinikerrostumat stroomassa | D: Hyaliinimateriaali värjäytyy kirkkaanpunaiseksi Masson-Trichrome |
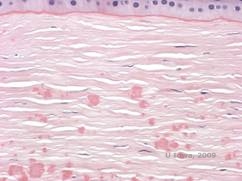 |
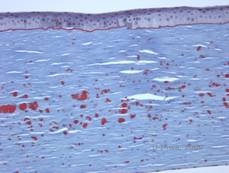 |
Granulaarinen sarveiskalvon dystrofia, tyyppi II
Granulaarinen sarveiskalvon dystrofia, tyyppi II (GCD2), joka aiemmin tunnettiin nimellä Avellino tai yhdistetty granulaarinen-ristikkomainen sarveiskalvon dystrofia, on autosomaalinen dominantti sairaus, joka liittyy TGFβI-geenin mutaatioon, joka johtaa sekä hyaliinin että amyloidin kerrostumiseen sarveiskalvon stroomassa. Tyypillisesti potilailla esiintyy toisella vuosikymmenellä pieniä harmaanvalkoisia pisteitä pinnallisessa stroomassa. Samentumat voivat myös näyttää piikkimäisiltä, rengasmaisilta tai tähtimäisiltä. Retrovalaistuksessa ne ovat osittain läpikuultavia. Myöhemmin tautiprosessissa niihin voi kehittyä myös ristikkoviivoja (ks. kuvat 4A ja 4B). Nämä viivat eivät risteä keskenään, ja ne näyttävät valkoisemmilta ja vähemmän taittuvilta kuin ristikkoviivat. GCD2:n oireita ovat kipu, johon liittyy epiteelin eroosioita ja näön heikkeneminen (2).
Histopatologisesti sarveiskalvolla on stroomakertymiä, jotka värjäytyvät punaisiksi Masson Trichrome -aineella osoittaen hyaliinin läsnäoloa (ks. kuva 4C). Lisäksi Kongon punaisella värjäytyminen osoittaa omenanvihreää kaksoissäröisyyttä ristipolarisaatiossa, mikä osoittaa amyloidin läsnäoloa (ks. kuva 4D). Taudin uskottiin saaneen alkunsa eräästä perheestä Avellinossa, Italiassa. GCD-tyypin II tautia on kuitenkin nyt raportoitu potilailla myös monista muista maista (2,5), ja suurin esiintyvyys on Itä-Aasiassa.
| A: Avellinon dystrofia, jossa näkyy ristikkomaisia ja granulaarisia kerrostumia sarveiskalvon stroomassa | B. Masson Trichrome -värjäys, joka osoittaa anteriorisia strooman hyaliinisia kerrostumia |
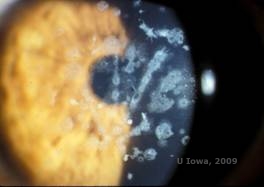 |
 |
| C: Kongon punavärjäys, joka osoittaa vaaleanpunaisia, amorfisia amyloidikertymiä samassa sarveiskalvon näytteessä. | D: Ristipolarisaatio paljastaa amyloidiin viittaavan omenanvihreän kaksoiskatkon. |
 |
 |
STROMAALINEN SYVÄKALVODYSTROFIA
MAKULAARINEN SYVÄKALVODYSTROFIA
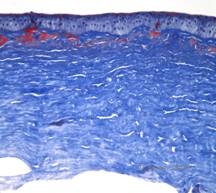
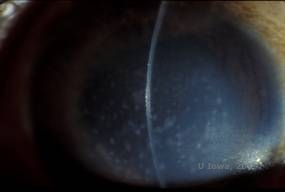
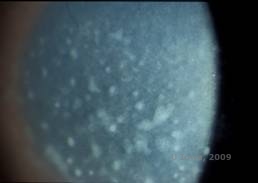
Makulaarinen sarveiskalvon dystrofia. (MCD) on autosomaalinen resessiivinen sairaus, joka johtuu kromosomissa 16 sijaitsevan hiilihydraattisulfotransferaasi 6 -geenin (CHST6) mutaatiosta, joka johtaa vikaan kerataanisulfaatin synteesissä, joka on sarveiskalvon tärkein glykosaminoglykaani. Se on harvinaisempi kuin LCD tai GCD, mutta sillä on taipumus vaikuttaa näkökykyyn vakavammin. Vaikka MCD on maailmanlaajuisesti harvinaisempi kuin LCD tai GCD, se on sarveiskalvon stroomadystrofioista yleisin esimerkiksi Islannissa ja Saudi-Arabiassa (2,6). GCD1:n kaltaisia harmaanvalkoisia, pilkullisia anteriorisia stroomavaurioita esiintyy sarveiskalvolla ensimmäisellä vuosikymmenellä. Toisin kuin GCD1:ssä, saostumien välissä on kuitenkin stroomaista sameutta, ja koko sarveiskalvo limbuksesta limbukseen on usein mukana (ks. kuvat 5A ja 5B). Sarveiskalvo on ohut, ja häiriön edetessä Descemetin kalvo harmaantuu ja siihen kehittyy guttia. Epiteelin eroosiota voi esiintyä, mutta vähemmän MCD:ssä kuin LCD:ssä. Potilaille kehittyy tyypillisesti vaikea näön heikkeneminen toiselle tai kolmannelle elinvuosikymmenelle mennessä sarveiskalvon diffuusin sameuden vuoksi. PTK voidaan suorittaa joissakin MCD:n varhaisissa tapauksissa. Tämä tila ei kuitenkaan yleensä sovellu PTK:lle yhtä hyvin kuin lattice- tai granulaarinen dystrofia, ja hoito edellyttää usein sarveiskalvonsiirtoa (7). Siirteiden uusiutuminen on MCD:ssä harvinaisempaa kuin granulaarisessa tai ristikkäisdystrofiassa (1,2,5,6,8).
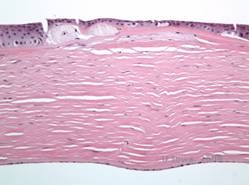
MCD:ssä stroomakertymät koostuvat mukopolysakkarideista, jotka kerääntyvät sarveiskalvon strooman keratosyyttien endoplasmiseen retikulumiin, ekstrasellulaarisesti stroomalamellien väliin sekä epiteelin, Descemet-kalvon ja endoteelin sisään. Nämä kerrostumat värjäytyvät siniseksi Alcian-sinisellä (ks. kuvat 5C ja 5D) (1). Bowmanin kerroksessa ja guttaeissa on katkoksia, ja Descemetin kalvo on paksuuntunut (2).
MCD:n kolme alatyyppiä on kuvattu immunoreaktiivisen kerataanisulfaatin esiintymisen tai puuttumisen perusteella eri kudoksissa. Tyypissä I ei ole immunoreaktiivista kerataanisulfaattia sarveiskalvon stroomassa, keratosyyteissä, seerumissa tai rustossa, ja se on maailmanlaajuisesti yleisin MCD:n muunnos. Tyypistä IA puuttuu kerataanisulfaattia stroomasta, seerumista ja rustosta, mutta sen pitoisuudet ovat havaittavissa keratosyyttien sisällä. Tyypin II kerataanisulfaattia esiintyy paljon pienempinä pitoisuuksina stroomassa, keratosyyteissä, seerumissa ja rustossa (6).
| A: Viiltolamppukuva makulan sarveiskalvon dystrofiasta. | B: Huomaa sarveiskalvon stroomakertymien välinen sameus |
 |
 |
| C: H&E sarveiskalvosta, jossa on makuladystrofia. Huomaa anterioriset stroomakertymät ja Bowmanin kerroksen häiriö | D: Keratosyyttien sisällä olevat mukopolysakkaridikerrostumat, jotka on korostettu Alcian Blue -värjäyksellä |
 |
 |
SCHNYDERIN SYRVÄKALVODYSTROFIA (SCD)
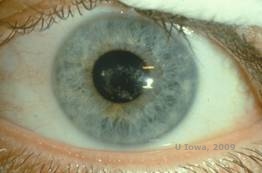
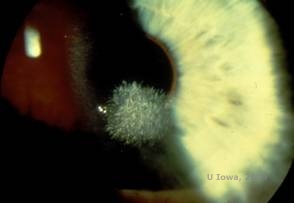
Schnyderin sarveiskalvodystrofia, joka tunnettiin aiemmin nimellä Schnyderin kiteinen sarveiskalvon dystrofia, on autosomaalisesti dominoiva, molemminpuolinen sarveiskalvon stroomadystrofia, joka liittyy geneettiseen mutaatioon kromosomissa 1 sijaitsevassa UbiA-prenyylitransferaasi-domeenia sisältävässä 1(UBIAD1)-geenissä. Tästä johtuva sarveiskalvon keratosyyttien aineenvaihduntahäiriö johtaa kiteisen kolesterolin kertymiseen stroomaan. Kiteiden esiintyminen ei kuitenkaan ole ehdottoman välttämätöntä SCD-diagnoosin tekemiseksi. Itse asiassa vain 54 prosentilla SCD-potilaista on sarveiskalvokiteitä. Tyypillisesti potilaat ilmaantuvat toisella tai kolmannella vuosikymmenellä, ja heillä on rengasmainen sarveiskalvon keskeinen sameus, jossa on tai ei ole pilkunmuotoisia subepiteliaalisia kiteitä (ks. kuvat 6A ja 6B). Arcus lipoides ilmaantuu 23-38 vuoden iässä. 38 ikävuoden jälkeen sarveiskalvon asteittainen samentuminen johtaa keskiperefääriin ulottuvaan panstromaaliseen sameuteen. Useimmilla yli 50-vuotiailla potilailla on valonäköhäiriöitä, häikäisyä ja heikentynyt sarveiskalvon tunto, minkä vuoksi he saattavat tarvita kirurgista hoitoa, kuten sarveiskalvonsiirtoa tai PTK:ta. Siirteessä voi esiintyä uusiutumista. Tauti on yhdistetty joillakin potilailla hyperkolesterolemiaan, hyperlipidemiaan ja genu valgumiin (2,5,9,10).
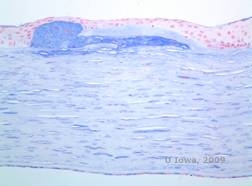
Histopatologisesti fosfolipideistä ja kolesterolista koostuvat kaksitahoiset kolesterolikiteet kerrostuvat tyvikalvon epiteelisolujen, keratosyyttien, Bowmanin kerroksen ja stroomalamellien väliin. Lipidit liukenevat normaalissa histologisessa käsittelyssä, joten sarveiskalvon läpi on otettava pakastepoikkileikkauksia, jotta lipidien esiintyminen voidaan osoittaa öljy-punapuna-O- tai sudanmustavärjäyksellä.
| A: Viiltolamppukuva Schnyderin sarveiskalvon dystrofiasta. | B. Keskellä sijaitsevat kiteiset kerrostumat |
 |
 |
| C: H&E sarveiskalvon H&E-kuva sarveiskalvon SCCD:stä | D. Oil Red O -värjäys korostaa kolesterolikiteitä, jotka näkyvät punaisina. |
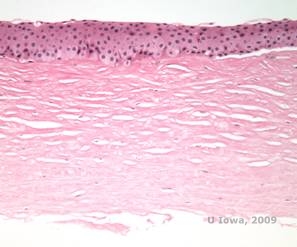 |
 |
Taulukkoon 4 on koottu yhteinen muistilista joidenkin stroomaan vaikuttavien sarveiskalvon dystrofioiden, niiden kerrostuman koostumuksen ja näiden kerrostumien värjäysmenetelmän muistamista varten.
Taulukko 4: Muistilista sarveiskalvon stroomadystrofioiden muistamista varten
- Marilyn-Macular Dystrophy
- Monroe-Mucopolysaccharide
- Always-Alcian Blue -värjäys
- Gets-Granular Dystrophy
- Her-Hyaline
- Man in-Masson Trichrome -värjäys
- Los-Lattice Dystrophy
- Angeles-Amyloid
- California-Congo Red
OVERVIEW: CORNEAL STROMAL DYSTROPHIES
EPIDEMIOLOGIA
|
MERKITSEVÄT
|
SYMPTOMIT
|
HOITO
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Sidekalvon ja sarveiskalvon patologia. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3. painos. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D classification of corneal dystrophies–edition 2. Cornea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. TGFBI-assosioituneiden sarveiskalvon dystrofioiden kliiniset ja geneettiset näkökohdat. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nature genet. 1994; 6: 47-51.
- Bron AJ. Sarveiskalvon dystrofioiden genetiikka: Mitä olemme oppineet viimeisten kahdenkymmenenviiden vuoden aikana. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetroiva keratoplastia makulan sarveiskalvon dystrofian hoidossa. Ophthalmology 2005;112: 220-224.
- Badr IA, Wagoner MD. Fototerapeuttinen keratektomia makulan sarveiskalvon dystrofian hoidossa. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetics of anterior and stromal corneal dystrophies. Seminars in Ophthalmology, 2008; 23: 1,9-17.
- Weiss JS. Schnyderin sarveiskalvon dystrofia. Curr Opin Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, ym. Schnyderin kiteisen sarveiskalvon dystrofian geeni karttuu ihmisen kromosomille 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Suggested citation format: Van C, Syed NA. Epiteelis-stroomaaliset ja stroomaaliset sarveiskalvon dystrofiat: A Clinicopathologic Review. Revision of ; EyeRounds.org. August 20, 2015. Saatavissa: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm