- Revizuirea clinico-patologică
- INTRODUCERE
- Distrofii corneene epiteliale-stromale
- DISTROFIA CORNEALĂ DE REIS-BUCKLERS
- DISTROFIE CORNEALĂ DE LATICE
- DISTROFIE CORNEALĂ GRANULARĂ, TIPUL I
- DISTROFIE CORNEALĂ GRANULARĂ, TIP II
- DISTROFIE CORNEALĂ STOMALĂ
- DISTROFIE CORNEALĂ MACULARĂ
- DISTROFIE CORNEALĂ SCHNYDER (SCD)
- Tabelul 4: Mnemotehnică pentru reamintirea distrofiilor stromale corneene
- EXAMINARE: DISTROFII CORNEALE STROMALE
- EPIDEMIOLOGIE
- SIGNE
- SIMPTOME
- TRATAMENT
Revizuirea clinico-patologică
Emily S. Birkholz, MD, Nasreen A. Syed, MD, și Michael D. Wagoner, MD, PhD
17 august 2009
Revizuirea majoră: Chaunhi Van, MD și Nasreen Syed, MD
August 20, 2015
INTRODUCERE
Distrofiile corneene epiteliale-stromale și stromale sunt un grup de afecțiuni ereditare ale corneei care sunt cauzate de acumularea progresivă de depozite în straturile corneei. Aceste depozite nu sunt cauzate de inflamații, infecții sau traume, ci de mutații genetice care duc la transcrierea unor proteine aberante, ceea ce duce la acumularea de material insolubil în interiorul corneei. Tulburările pot afecta sau nu vederea și pot fi sau nu simetrice (1). Sistemul de clasificare din 2015 al Comitetului internațional pentru clasificarea distrofiilor corneene (IC3D) a împărțit distrofiile corneene în 4 categorii: distrofii epiteliale și subepiteliale, distrofii epiteliale-stromale, distrofii stromale, distrofii stromale și distrofii endoteliale. Majoritatea distrofiilor considerate anterior stromale sunt acum clasificate fie ca distrofii epiteliale-stromale, fie ca distrofii stromale. În tabelele 1 și 2 sunt enumerate distrofiile epiteliale-stromale și distrofiile stromale (2). Vechea clasificare pentru distrofiile stromale corneene este prezentată în tabelul 3.
- Distrofia corneană Reis-Bucklers
- Distrofia corneană Thiel-Behnke
- Distrofia corneană reticulară
- Distrofia corneană reticulară, tip 1 și variante
- Distrofie corneană granulară, tip 1
- Distrofie corneană granulară, tip 2
- Distrofia corneană maculară
- Distrofia corneană Schnyder
- Distrofia corneană stromală stromală congenitală
- Distrofia corneană amorfă posterioară
- Distrofia corneană noroasă centrală a lui Francois
- Distrofia corneană pre-Distrofia corneană Descemet
Tabelul 3. Vechea clasificare a distrofiilor stromale corneene
- Distrofia corneană cu rețea
- Distrofia corneană granulară
- Distrofia corneană Avellino
- Distrofia corneană maculară
- Distrofia corneană gelatinoasă-.like dystrophy
- Schnyder corneal dystrophy
- Francois-Neetans Fleck dystrophy
- Distrofia stromală ereditară congenitală ereditară
Distrofii corneene epiteliale-stromale
Distrofiile epiteliale-stromale sunt cauzate de mutații în gena indusă de factorul de creștere transformant beta (TGFβI), cunoscută și sub numele de gena BIGH3. TGFβI este localizată pe cromozomul 5q31 și codifică pentru keratoepitelină, o proteină secretată de epiteliul corneei. Această proteină acționează ca o proteină de aderență și este prezentă în stroma normală. Fiind o proteină mică, aproximativ de mărimea albuminei, are capacitatea de a difuza prin stroma corneană. Atunci când apare o mutație în gena TGFβI, structura keratoepitelinei este anormală și are loc o acumulare a proteinei insolubile sau a fragmentelor sale proteolitice în cornee (1, 3). Este interesant faptul că mutația genei TGFβI a fost descoperită în parte la Universitatea din Iowa. Un grup de cercetători și clinicieni, printre care Edwin M. Stone, Robert Folberg și Jay H. Krachmer, au cartografiat distrofia granulară de tip I, distrofia granulară de tip II și distrofia reticulară pe cromozomul 5q în 1994 (4). Până în prezent, au fost identificate 63 de mutații diferite în gena TGFβI. Nu au fost identificate tratamente eficiente pentru a preveni sau atenua depunerea keratoepitelinei. Distrofiile au de obicei o moștenire autosomal dominantă și implică stratul Bowman și stroma (3).
DISTROFIA CORNEALĂ DE REIS-BUCKLERS
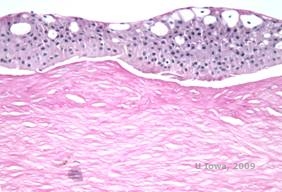
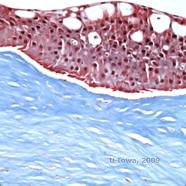
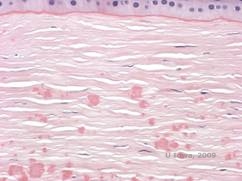
Reis-Bücklers, cunoscută anterior ca distrofia corneană granulară de tip III sau distrofia corneană de tip I a lui Bowman, prezintă de obicei cornee normale la naștere, dar dezvoltă eroziuni recurente dureroase, opacizare și pierderea progresivă a vederii în primul deceniu de viață (1). Opacitățile neregulate, alb-cenușii, de tip geografic, sunt localizate în stratul Bowman și în stroma anterioară. În stadiile mai avansate ale bolii, opacitățile se pot extinde la limbus și la stroma mai profundă (2). Histopatologia relevă depozite anterioare stromale și subepiteliale de material asemănător cu hialina, care perturbă și adesea înlocuiesc stratul lui Bowman (a se vedea figura 1A și 1B). Depozitele se colorează în roșu cu colorația tricromă Masson (2). Din punct de vedere ultrastructural, materialul de tip hialină constă în corpuri asemănătoare unor tije, ceea ce ajută la distingerea de distrofia corneană Thiel-Behnke (1, 2).
| A: H&E de Reis Bückler care arată distrugerea stratului lui Bowman și epiteliul neregulat | B. Colorație Masson Trichrome demonstrând colorația epitelială |
 |
 |
DISTROFIE CORNEALĂ DE LATICE
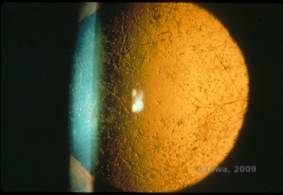
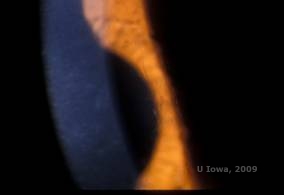
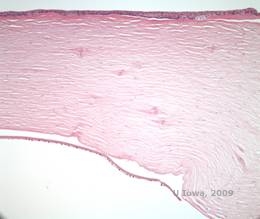
Distrofia corneană de latice (LCD) este cea mai frecventă dintre distrofiile epiteliale-stromale ale corneei. Este de obicei o boală autosomal dominantă, bilaterală, care se prezintă de obicei spre sfârșitul primului deceniu de viață cu simptome de eroziuni corneene recurente și scăderea vederii. Se caracterizează prin linii de zăbrele care sunt opacități refractile liniare, orientate radial, ramificate, descrise ca fiind „ca de sticlă”, localizate în stroma anterioară (a se vedea figura 2A și 2B). Aceste linii reticulare se găsesc inițial în corneea centrală superficială. Pe măsură ce boala progresează, ele se răspândesc mai adânc și periferic în stromă, cu cruțarea limbusului (1, 2). Alte constatări la examen includ opacități asemănătoare unor pete, puncte albe subepiteliale și opacitate stromală „ground-glass”, care începe central și devine mai difuză (2). Mulți pacienți cu LCD vor avea nevoie de intervenție chirurgicală pentru tratarea eroziunilor recurente și a scăderii vederii. În cazul în care boala este localizată anterior în stromă, pacienții pot fi adesea tratați cu succes prin keratectomie fototerapeutică (PTK). Unii necesită transplant de cornee. Deoarece keratoepitelina, proteina produsă de gena TGFβI, este produsă mai ales în epiteliul corneei, boala tinde să recidiveze în grefele de cornee (1).
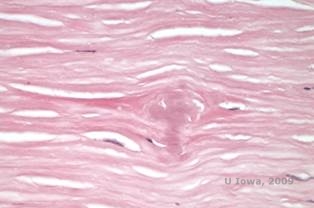
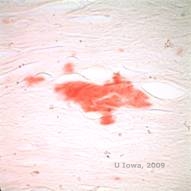
În LCD, depozitele de amiloid se acumulează între membrana bazală epitelială și stratul Bowman, precum și în stromă, provocând o distorsiune a arhitecturii lamelare. Depozitele se colorează pozitiv la imunohistochimie folosind anticorpi împotriva keratoepitelinei (2). Depozitele apar ca depozite amorfe de culoare roz la colorația cu hematoxilină și eozină (H&E) (a se vedea figura 1C și 1D) și se colorează cu colorația cu roșu Congo, demonstrând birefringența clasică de culoare verde măr la polarizarea încrucișată (a se vedea figura 2E și 2F) (1). Absența sau subțierea stratului Bowman, atrofia epitelială și degenerarea epitelială bazală pot fi, de asemenea, constatate la histopatologie în LCD (2).
LCD tip I este forma clasică de LCD cauzată de o mutație în gena TGFβI care are ca rezultat depunerea izolată de amiloid în cornee. Au fost identificate patru variante de LCD: LCD tip IIIA, tip I/IIIA, tip IV și amiloidoza polimorfă. Variantele LCD se prezintă mai târziu în viață decât LCD clasic. LCD tip IIIA se prezintă în decada a 5-a-7-a, de obicei cu eroziuni epiteliale. Are linii de grilaj mai groase, descrise ca „cu aspect de rozătoare”, care se extinde până la limbus. LCD tip I/IIIA are linii de rețea subțiri. LCD de tip IV se prezintă în decada a 7-a-9-a, cu linii reticulare mici. Depozitele de amiloid în LCD tip IV se găsesc în stroma profundă și rareori apar eroziuni epiteliale. Liniile de zăbrele sunt absente în amiloidoza de tip polimorf și rareori apar eroziuni epiteliale (2).
LCD tip II este un sindrom de amiloidoză sistemică cunoscut sub numele de sindrom Meretoja care afectează pielea, nervii cranieni și corneea. Se prezintă la vârsta adultă timpurie cu neuropatii periferice, neuropatii craniene, facies de câine de vânătoare, piele uscată, blefarochalasis, buze proeminente și linii de zăbrele corneene. Acest tip a fost legat de gena gelsolin de pe cromozomul 9, care codifică pentru o proteină precursoare amiloidă care are rolul de a elimina actina din locurile de leziune și inflamație (1). Denumirea este o denumire greșită și nu este considerată o variantă a distrofiei corneene în rețea (2).
| A: Ochiul stâng pe retroiluminare care demonstrează depozitele stromale anterioare în distrofia corneană în rețea | B: Ochiul stâng cu putere mai mare care arată depozitele stromale anterioare liniare. |
 |
 |
| C: Colorație H&E a corneei cu rețea. Se observă depozitele amorfe roz din stromă | D: O vedere mai apropiată a depozitelor roz, amorfe |
 |
 |
| E: Colorație cu roșu Congo, evidențiind amiloidul | F: Birefringența verde-măr a amiloidului cu polarizare încrucișată. |
 |
 |
DISTROFIE CORNEALĂ GRANULARĂ, TIPUL I
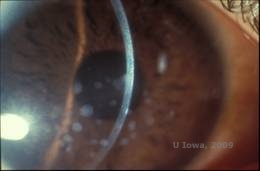
Distrofie corneană granulară, tip I (GCD1) este o boală bilaterală, autosomal dominantă, asociată cu o mutație în gena TGFβI care duce la depunerea unui material hialin în stroma corneană. De obicei, se prezintă la începutul primului deceniu de viață cu opacitate alb-cenușie, de tip „firimitură”, în stroma anterioară și mijlocie, extinzându-se în stroma posterioară în boala avansată (1, 2). Aceste opacități sunt depozite discrete localizate central, cu cornee clară localizată la periferie și cornee clară între depozite (a se vedea figura 3A și 3B). Boala este de obicei asimptomatică la început, dar cu timpul opacitățile se pot coagula și pot duce la scăderea vederii. Erodările corneene recurente pot apărea în GCD, dar cu o incidență mai mică decât în LCD (1, 5). Pacienții pot prezenta, de asemenea, orbire și fotofobie (2). Tratamentul la începutul procesului bolii este adesea doar observația. Cu toate acestea, pe măsură ce boala progresează, PTK și transplantul de cornee pot fi necesare pentru a îmbunătăți vederea și simptomele de eroziune. Ca și în cazul LCD, boala poate recidiva în grefele de cornee.
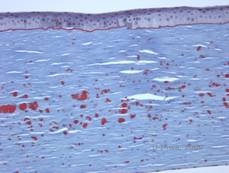
Histopatologic, opacitățile sunt depozite eozinofile descrise adesea ca fiind „ca bomboanele de ciocolată” în stroma anterioară, alcătuite dintr-un material asemănător cu hialina. Cu timpul, depozitele progresează în stroma corneană mai profundă. Materialul hialin se colorează în roșu aprins cu colorația trichromă Masson (vezi figurile 3C și 3D).
| A:Fotografie cu lampă cu fantă a distrofiei corneenei granulare, tip I | B: Observați depozitele stromale „ca niște firimituri” cu stromă intermediară clară. |
 |
 |
| C: Colorația H& E a corneei care arată depozite hialine eozinofile „ca o bomboană de ciocolată” în stromă | D: Materialul hialin se colorează în roșu aprins cu Masson-Trichrome |
 |
 |
DISTROFIE CORNEALĂ GRANULARĂ, TIP II
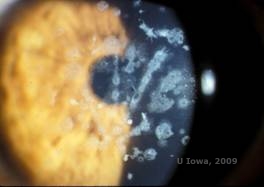
Distrofie corneană granulară, tip II (GCD2), cunoscută anterior sub numele de distrofia corneei Avellino sau distrofia corneană combinată granulară cu rețea, este o boală autosomală dominantă legată de o mutație în gena TGFβI care duce la o depunere atât de hialină, cât și de amiloid în stroma corneană. În mod obișnuit, pacienții se prezintă în al doilea deceniu de viață cu mici puncte alb-cenușii în stroma superficială. Opacitățile pot avea, de asemenea, o formă spinoasă, inelară sau stelată. În retroiluminare, acestea sunt parțial translucide. Mai târziu, în procesul bolii, ele pot dezvolta și linii reticulare (a se vedea figura 4A și 4B). Aceste linii nu se încrucișează între ele și apar mai albe și mai puțin refractile decât liniile reticulare. Simptomele GCD2 sunt durerea cu eroziuni epiteliale și tulburări de vedere (2).
Histopatologic, corneea va avea depozite stromale care se colorează în roșu cu Masson Trichrome, indicând prezența hialinei (Vezi Figura 4C). În plus, colorația cu roșu Congo va demonstra birefringență verde-măr la polarizarea încrucișată, indicând prezența amiloidului (A se vedea figura 4D). Se crede că această boală provine de la o familie din Avellino, Italia. Cu toate acestea, GCD tip II a fost acum raportată și la pacienți din multe alte țări (2,5), cea mai mare prevalență fiind în Asia de Est.
| A: Distrofia Avellino care prezintă depozite de tip rețea și depozite de tip granular în stroma corneană | B. Colorație Masson Trichrome demonstrând depozite hialine în stromul anterior |
 |
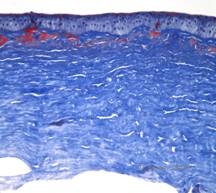 |
| C: Colorație cu roșu Congo care arată depozite amiloide roz, amorfe, în același specimen cornean. | D: Polarizarea încrucișată relevă birefringența verde măr care indică amiloid. |
 |
 |
DISTROFIE CORNEALĂ STOMALĂ
DISTROFIE CORNEALĂ MACULARĂ
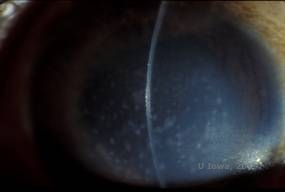
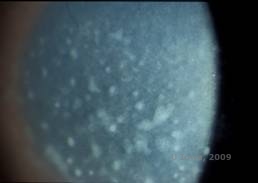
Distrofie corneală maculară. (MCD) este o boală autozomal recesivă cauzată de o mutație în gena carbohidrat sulfotransferazei 6 (CHST6) de pe cromozomul 16 care duce la un defect în sinteza de keratan sulfat, principalul glicozaminoglican al corneei. Este cea mai puțin frecventă decât LCD sau GCD, dar tinde să aibă un impact mai sever asupra vederii. Deși MCD este mai puțin frecventă la nivel mondial decât LCD sau GCD, este cea mai frecventă dintre distrofiile stromale corneene în locuri precum Islanda și Arabia Saudită (2,6). Leziunile stromale anterioare de culoare alb-cenușie, asemănătoare cu GCD1 apar în cornee în primul deceniu de viață. Spre deosebire de GCD1, însă, există o ceață stromală între depozite și întreaga cornee, de la limbus la limbus, este adesea implicată (a se vedea figura 5A și 5B). Corneea este subțire și, pe măsură ce tulburarea progresează, membrana Descemet devine gri și dezvoltă guttae. Pot apărea eroziuni epiteliale, dar mai puțin în MCD decât în LCD. Pacienții dezvoltă de obicei o pierdere severă a vederii până în a doua sau a treia decadă a vieții din cauza ceții corneene difuze. PTK poate fi efectuată în unele cazuri timpurii de MCD. Cu toate acestea, această afecțiune nu se pretează, în general, la PTK la fel de bine ca distrofia reticulară sau granulară și necesită adesea transplant cornean pentru tratament (7). Recidiva în grefe este mai puțin frecventă în cazul MCD decât în cazul distrofiei granulare sau lattice (1,2,5,6,8).
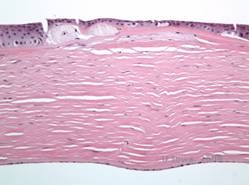
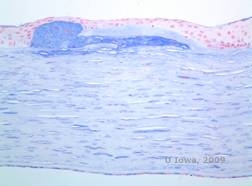
Depozitele stromale în MCD sunt compuse din mucopolizaharide care se acumulează în reticulul endoplasmatic al keratocitelor din stroma corneană, extracelular între lamelele stromale și în epiteliu, membrana Descemet și endoteliu. Aceste depozite se colorează în albastru cu albastru Alcian (a se vedea figura 5C și 5D) (1). Există rupturi în stratul Bowman și guttae cu îngroșarea membranei Descemet (2).
Au fost descrise trei subtipuri de MCD pe baza prezenței sau absenței de keratan sulfat imunoreactiv în diferite țesuturi. Tipul I nu prezintă keratan sulfat imunoreactiv în stroma corneană, keratocite, ser sau cartilaj și este cea mai frecventă variantă de MCD la nivel mondial. Tipul IA este lipsit de keratan sulfat în stromă, ser și cartilaj, dar are niveluri detectabile în interiorul keratocitelor. Tipul II are keratan sulfat prezent la niveluri mult reduse în stromă, keratocite, ser și cartilaj (6).
| A: Fotografie cu lampă cu fantă a distrofiei maculare corneene. | B: Observați ceața dintre depozitele stromale corneene |
 |
 |
| C: H&E de cornee cu distrofie maculară. Observați depozitele stromale anterioare și întreruperea stratului lui Bowman | D: Depozite de mucopolizaharide în interiorul keratocitelor evidențiate cu colorare cu albastru Alcian |
 |
 |
DISTROFIE CORNEALĂ SCHNYDER (SCD)
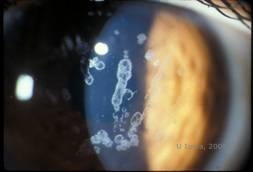
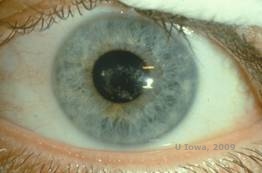
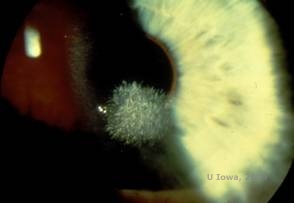
Distrofie corneană Schnyder (SCD), cunoscută anterior sub numele de distrofie corneană cristalină Schnyder, este o distrofie stromală corneană bilaterală, autozomal dominantă, legată de o mutație genetică în gena UbiA preniltransferaza care conține domeniul 1 (UBIAD1) de pe cromozomul 1. Defectul metabolic rezultat al keratocitelor corneene duce la depunerea de colesterol cristalin în stromă. Cu toate acestea, prezența cristalelor nu este absolut necesară pentru diagnosticul de SCD. De fapt, doar 54% dintre pacienții cu SCD au cristale corneene. În mod obișnuit, pacienții se prezintă în a doua sau a treia decadă cu opacitate corneană centrală în formă de inel, cu sau fără cristale subepiteliale în formă de virgulă (a se vedea figura 6A și 6B). Apoi, arcul lipoidian apare între 23 și 38 de ani. După vârsta de 38 de ani, opacifierea corneană progresivă are ca rezultat o ceață panstromală care ajunge până la jumătatea periferiei. Majoritatea pacienților cu vârsta de peste 50 de ani prezintă pierdere a vederii fotopice, orbire și diminuarea senzației corneene și, prin urmare, pot necesita tratament chirurgical, inclusiv transplant cornean sau PTK. Pot apărea recidive în grefă. Boala a fost asociată cu hipercolesterolemia, hiperlipidemia și genu valgum la unii pacienți (2,5,9,10).
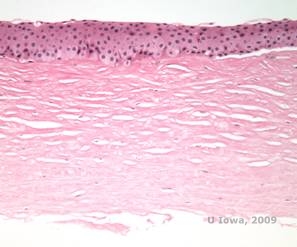
Histopatologic, cristale de colesterol birefringente compuse din fosfolipide și colesterol se depun în interiorul celulelor epiteliale bazale, keratocite, stratul lui Bowman și între lamelele stromale. Lipidele se dizolvă în procesarea histologică normală, astfel încât trebuie obținute secțiuni congelate prin cornee pentru a demonstra prezența lipidelor cu ajutorul colorațiilor Oil-Red-O sau Sudan black.
| A: Fotografie cu lampă cu fantă a distrofiei corneenei Schnyder. | B. Depozite cristaline localizate central |
 |
 |
| C: H&E de cornee cu SCCD | D. Colorația Oil Red O evidențiază cristalele de colesterol care apar roșii. |
 |
 |
Tabelul 4 oferă un mnemotehnic comun pentru memorarea unora dintre distrofiile corneene care afectează stroma, se enumeră compoziția depozitului lor și metoda de colorare a acestor depozite.
Tabelul 4: Mnemotehnică pentru reamintirea distrofiilor stromale corneene
- Distrofia Marilyn-Maculară
- Monroe-Mucopolizaharide
- Colorația Always-Alcian Blue
- Gets-Granular Dystrophy
- Her-Hyaline
- Man in-Masson Trichrome stain
- Los-Lattice Dystrophy
- Angeles-Amyloid
- California-Congo Red
EXAMINARE: DISTROFII CORNEALE STROMALE
EPIDEMIOLOGIE
|
SIGNE
|
SIMPTOME
|
TRATAMENT
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Patologia conjunctivală și corneană. Albert and Jakobiec’s Principles and Practice of Ophthalmology, ediția a 3-a. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3rd edition. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D classification of corneal dystrophies–edition 2. (Clasificarea IC3D a distrofiilor corneene – ediția 2). Cornea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. Clinical and genetic aspects of the TGFBI-associated corneal dystrophies. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nature genet. 1994; 6: 47-51.
- Bron AJ. Genetica distrofiilor corneene: Ce am învățat în ultimii douăzeci și cinci de ani. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Keratoplastia penetrantă pentru distrofia corneană maculară. Ophthalmology 2005;112: 220-224.
- Badr IA, Wagoner MD. Keratectomia fototerapeutică pentru distrofia corneană maculară. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetica distrofiilor corneene anterioare și stromale. Seminars in Ophthalmology, 2008; 23: 1,9-17.
- Weiss JS. Distrofia corneană Schnyder. Curr Opin Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. The gene for Schnyder’s crystalline corneal dystrophy maps to human chromosome 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Formatul de citare sugerat: Van C, Syed NA. Distrofii corneene epiteliale-stromale și stromale: A Clinicopathologic Review. Revizuire a ; EyeRounds.org. 20 august 2015. Disponibil la: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm