- Fundo
- Fisiologia normal e fisiologia patofisiológica do potássio
- Potencial de ação de um cardiomiócito não-pacemaker
- Potencial de ação de uma célula de marcapasso cardíaco
- Condução de corrente
- Período refratário
- Hipercalemia, classificação e causas
- Classificação
- Causas
- Efeitos da hipercalemia
- Efeitos metabólicos
- Bomba de potássio de sódio
- Conclusão
Fundo
Potássio é um cátion macio, branco-prateado altamente reativo pertencente à família dos metais alcalinos na tabela periódica. É o cátion mais abundante no corpo humano como um todo, e o íon mais difundido em seus compartimentos intracelulares.
Em média, uma dieta ocidental contém de 80-100 mEq de potássio por dia, e sob condições fisiológicas normais, 90% dele é absorvido passivamente, deixando apenas 9,0 mmol para excreção fecal. Os 3500-4000 mmol armazenados no corpo são desproporcionais aos níveis de potássio do plasma diurno que são normalmente mantidos na faixa de 3,5-5,3 mmol/L através de um mecanismo de homeostase apertado com os níveis mais baixos sendo à noite e nas primeiras horas da manhã e o nível mais alto de pico nas horas da tarde.
Após absorvido na corrente sanguínea, torna-se o papel do rim adequar a ingestão de potássio ao débito de potássio; requerendo várias horas, durante as quais o “equilíbrio interno de potássio” sob a influência da insulina e das catecolaminas mantém a homeostase temporária, deslocando o potássio entre os espaços intracelular e extracelular. A estimulação dos receptores alfa dificulta a entrada de potássio nas células, e a estimulação dos receptores beta o promove ativando a bomba de sódio potássico ATPase.
A bomba de sódio-potássio ATPase é a enzima gate-keeper localizada no sarcolemma. Ela ajuda a salvaguardar 98% do potássio (aproximadamente 144,0 mmol) retido no interior da célula. Isto assegura a preservação da diferença de potencial vital através das membranas celulares necessárias para o funcionamento celular adequado, especialmente as células excitáveis, como as células nervosas e as células musculares cardíacas.
Fisiologia normal e fisiologia patofisiológica do potássio
Após sua rápida absorção, o potássio ajuda a orquestrar seus próprios níveis corporais através da liberação de insulina e aldosterona. Outros estímulos corporais inerentes também encontrados para controlar os níveis corporais de potássio incluem receptores beta-2 adrenérgicos, PH sanguíneo alcalino, e anabolismo celular.
Lançamento da Insulina e Aldosterona: O potássio ingerido entra rapidamente na circulação. Ao chegar à circulação portal, estimula o pâncreas a liberar insulina. Ao mesmo tempo, o potássio circulante que chega às células justa aglomerulares resulta na libertação de renina. A renina, ao alcançar o fígado, é convertida em angiotensina I. A angiotensina I viaja para os pulmões onde é convertida em angiotensina II. A angiotensina II completa então a sua viagem de volta aos rins através do sangue circulante para estimular a zona glomerulosa a secretar aldosterona.
Equilíbrio Potássico Interno: A insulina liberada pós-prandialmente atua principalmente sobre os músculos esqueléticos, ativando duas vias, a via dependente de AKT responsável pela inserção do GLUT4 transportador de glicose e a via APK ativando a ATPase celular de sódio potássio para deslocar o potássio para o espaço intracelular. Ao contrário da via dependente de AKT, a via APK não é prejudicada pela síndrome metabólica nem por doença renal crônica (Figura 1).
Excreção: O potássio filtrado pelos glomérulos renais é reabsorvido passivamente no túbulo proximal e no laço de Henle em proporção à quantidade de sódio e água fornecida. Normalmente apenas cerca de 10% da carga filtrada atinge o nefrónio distal.
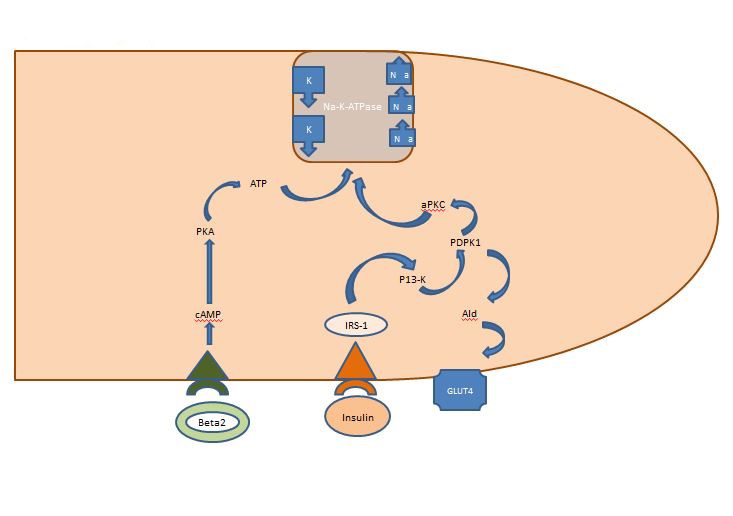
Figure 1. Acção da insulina sobre uma célula muscular esquelética. A insulina liberada pós-prandialmente ativa duas vias nos músculos esqueléticos, a via dependente de AKT responsável pela inserção do GLUT4 transportador de glicose e a via APK ativando a ATPase sódica celular potássica para deslocar o potássio para o espaço intracelular.
No início do túbulo convoluto distal, a secreção de excesso de potássio começa e aumenta progressivamente à medida que avança mais para o nefrónio distal e para o ducto coletor. Isto é mediado pela upregulação do hidrogênio potássico ATPase sobre as células alfa-intercaladas.
Na hipercalemia, a quota de potássio excretado através do cólon pode aumentar em até 30%, por exemplo, em casos de insuficiência renal, onde o potássio é então activamente absorvido pela bomba de sódio activada ATPase de potássio na membrana basolateral do enterócito colónico, para ser excretado do outro lado, na luz do cólon através dos grandes canais de potássio apicais dependentes de cálcio das células.
É assim discernível do exposto acima que o mecanismo da homeostase do nível plasmático de potássio é ordenado principalmente pela interação de três transações simultâneas – ingestão de potássio, turnos intra/extracelulares de potássio e excreção urinária de potássio, todos os quais dependem em última instância da bomba de potássio de sódio.
Para compreender o mecanismo de perigo iminente da hipercalemia e seu manejo, deve-se compreender a fisiologia do potencial de ação e as entranhas da enzima ATPase sódio potássica.
Electrofisiologia do potencial de ação, ou seja movimento iônico através das membranas celulares, é determinado pela diferença em dois potenciais, um “potencial químico” no qual os íons se movem para baixo em seu gradiente de concentração e um “potencial elétrico” no qual íons e moléculas se repelem como cargas, produzindo o potencial transmembrana (TMP), que se diz ser +ve quando o movimento líquido de +ve íons é para o exterior da célula e vice-versa.
Potencial de ação de um cardiomiócito não-pacemaker
Existem cinco fases para um potencial de ação, que começam e terminam na fase 4. As bombas envolvidas neste processo incluem o sarcolemma permutador de cálcio sódico, a ATPase cálcica e, por fim, a ATPase sódica potássica.
- Fase 4. A fase de repouso: esta tem um potencial de repouso de -90 mV como resultado do movimento constante do potássio para fora através dos canais retificadores de entrada. Durante esta fase, os canais de sódio e cálcio são fechados.
- Fase 0. A fase de despolarização: a queima de uma célula de marcapasso ou sua condução através de uma célula vizinha desencadeia a elevação da TMP para acima de -90 mV. Neste ponto, os “canais rápidos de sódio” começam a abrir um a um, permitindo que o sódio entre na célula, elevando a TMP e, uma vez que os canais rápidos de sódio tenham se aberto o suficiente para render -70 mV, uma corrente interna de sódio auto-sustentada é colocada em movimento, despolarizando rapidamente o TMP a 0 mV para um provisório transitório conhecido como “overhoot”, momento em que os canais de sódio rápidos dependentes do tempo se fecham e os canais de cálcio de “abertura longa” se abrem para elevar o TMP a -40 mV e permitir um pequeno influxo constante de cálcio para baixo do seu gradiente de concentração.
- Fase 1. A fase inicial de repolarização: começa com o TMP ligeiramente +ve e a breve abertura de alguns canais de potássio resultando em seu fluxo para o exterior da célula, retornando o TMP para aproximadamente 0 mV.
- Fase 2. A fase de platô: aqui as duas correntes contrárias são balanceadas eletricamente e resultam na manutenção do TMP balanceado a pouco menos de 0 mV. “Os canais de cálcio de abertura longa” ainda estão abertos, resultando em um fluxo constante de cálcio para dentro da célula. O canal retificador retardado de potássio permite a passagem de potássio para o exterior da célula, diminuindo o seu gradiente de concentração.
- Fase 3. A fase de repolarização: durante esta fase, os canais de cálcio são gradualmente inativados e o fluxo persistente de potássio para o exterior da célula excede assim o fluxo interno de cálcio, devolvendo o potássio ao espaço intracelular e o sódio e o cálcio ao exterior da célula.
Potencial de ação de uma célula de marcapasso cardíaco
As células de marcapasso cardíaco têm uma automaticidade inata, permitindo sua despolarização em ciclos rítmicos. O nó sinoatrial (SAN) tem o maior ritmo de despolarização auto-iniciada a uma taxa de 60-90/min, seguido pelo nó atrioventricular (AVN) a uma taxa de 40-60/min e depois as fibras de Purkinje e o músculo ventricular a 20-40/min.
Os potenciais de membrana das células do marcapasso são instáveis e seus potenciais de ação não têm fases claras. Elas têm menos canais retificadores internos de potássio e seu TMP nunca cai abaixo de -60 mV, eliminando o papel dos canais rápidos de sódio que requerem um TMP de -90 mV resultando na ausência da fase de despolarização rápida.
Na TMP >-60 mV, a corrente “funny/pacemaker” é colocada em ação com um fluxo espontâneo de íons através dos canais lentos de sódio, despolarizando a TMP para <-50 mV e depois de volta para -60 mV quando os canais de cálcio se fecham.
Condução de corrente
Todos os cardiomiócitos são acoplados eletricamente através da junção da fenda, incluindo a célula do marcapasso. Isto facilita a despolarização generalizada de todas as células vizinhas, transformando o coração em uma unidade funcional na qual a célula com maior taxa inerente se torna o “marcapasso”.
Período refratário
O período refratário mais longo durante o longo platô na fase 2 devido aos canais lentos de cálcio proporciona o tempo necessário para o esvaziamento completo dos ventrículos antes da próxima contração. Os períodos refratários podem ser absolutos (ARP), efetivos (ERP) ou relativos (PRR). Em uma ARP, a célula é absolutamente insuperável.
Uma ERP dura desde a ARP até o segmento curto da fase 3. Um estímulo neste ponto poderia minimizar a despolarização da célula, mas o nível de despolarização é mais fraco do que a propagação de um potencial de ação para as células vizinhas.
RRP é trazido por um estímulo acima do normal, levando à despolarização da célula e à produção de um potencial de ação.
Um “período supra-normal” é um estado hiperexcitável durante o qual um estímulo mais fraco que o normal poderia levar a uma arritmia, necessitando da sincronização durante a cardioversão para evitar a fibrilação ventricular (Figura 2).
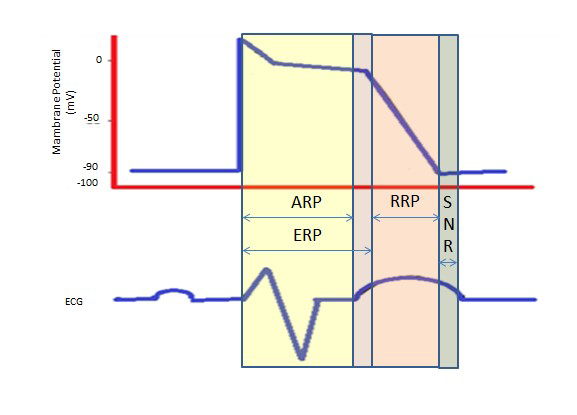
Figure 2. Períodos Refratários. ARP: Período Absoluto Refratário; ERP: Período Refratário Efetivo; RRP: Período Refratário Relativo; SNR: Período Refratário Supranormal
Hipercalemia, classificação e causas
>
Classificação
>
Hipercalemia é classificada como leve quando os níveis estão na faixa de 5,5-6,0 mmol/L, moderada de 6,1-6,9 mmol/L e severa em níveis de 7.0 mmol/L ou maior, e em qualquer nível em que ocorram alterações do ECG .
Causas
Hipercalemia ocorre quando os mecanismos compensatórios não são mais capazes de lidar com o desequilíbrio, razão pela qual geralmente é multifatorial.
- Incremento na ingestão de potássio por qualquer via, por exemplo, ingestão oral dietética, ou administração intravenosa de potássio contendo líquidos como a penicilina G.
- Retenção pelos rins: já que a excreção de potássio depende da aldosterona e do fornecimento de uma quantidade distal suficiente de sódio e água dentro dos nefrónios, condições como insuficiência renal, insuficiência adrenal (doença de Addison) , hipoaldosteronismo hiporeninémico tipo IV, A acidose tubular renal, especialmente em pacientes com nefropatia diabética, assim como qualquer condição que promova hipoperfusão como em esgotamento de volume e insuficiência cardíaca congestiva, afetará o intricado equilíbrio de potássio no organismo e predisporá à hipercalemia.
- Insuficiência renal: esta deve ser excluída em pacientes hipercalêmicos, particularmente na presença de hiponatremia e fraqueza muscular. Para triagem da Insuficiência Adrenal primária, é realizado um teste padrão de estimulação de cosyntropin, no qual 0,25 mg de cosyntropin sintético é administrado como um bolus intravenoso seguido pela medição do cortisol plasmático 45 minutos a 1 hora depois. Valores inferiores a 20 mcg/dL sugerem insuficiência adrenal.
- Drogas que retêm potássio: medicamentos prescritos que reduzem a atividade da ATPase sódica potássica, como bloqueadores dos receptores beta-adrenérgicos, e drogas que reduzem a secreção de aldosterona, como inibidores da ECA e da BRA, antiinflamatórios não-esteróides e diuréticos com supressão de potássio, necessitam de acompanhamento próximo para evitar a hipercalemia iatrogênica, especialmente na faixa etária geriátrica, com declínio progressivo da função renal como parte do processo de envelhecimento.
- Perturbações no deslocamento transcelular do potássio: pode ocorrer com condições de acidose, hiperglicemia, hiperosmolalidade, exercício severo, ruptura tecidual, paralisia periódica hipercalêmica e com bloqueadores beta-adrenérgicos. Para cada diminuição de 0,1 unidade no pH sanguíneo, o potássio sérico aumenta cerca de 0,6 mmol/L (menos se a acidose for causada por ácidos orgânicos) .
- Pseudo-hipoaldosteronismo é uma doença autossômica recessiva congênita na qual os rins são resistentes às ações da aldosterona.
- Pseudo-hipercalemia também não deve ser negligenciada: como o nome indica, isto ocorre quando há elevação do potássio sérico na presença de potássio plasmático normal. Pode ser visto em sangue hemolisado, torniquete apertado prolongado durante um procedimento de coleta de sangue, causando a liberação extracelular de potássio, com cerramento repetido do punho durante flebotomia, punção venosa traumática, com leucocitose e trombocitose, e em algumas síndromes genéticas incomuns como pseudo-hiperalemia familiar e esferocitose hereditária. Entretanto, pode ser simplesmente resultado de um simples erro laboratorial.
Efeitos da hipercalemia
A hipercalemia leve é frequentemente assintomática, detectada acidentalmente por testes laboratoriais, devido aos seus sintomas vagos como mal-estar, fraqueza muscular e paraestesia. A hipercalemia grave afetará a função neuromuscular na forma de fraqueza muscular esquelética e paralisia; entretanto, esta não é uma apresentação freqüente, pois a toxicidade cardíaca domina o quadro e é a apresentação preliminar. A toxicidade cardíaca normalmente se apresentará no ECG de forma escalonada, embora não necessariamente, dependendo da etiologia:
- Em níveis superiores a 5,5 mEq/L, o aumento da condutância dos canais de potássio aumenta a corrente lkr, levando a uma rápida repolarização na forma de uma onda T de pico na superfície do ECG. Estas ondas T podem ser diferenciadas das ondas de infarto do miocárdio e AVC por sua curta duração variando de 150-250 msec.
- Em níveis de potássio superiores a 6,5 mEq/L, ocorre um estado de despolarização sustentada do suben- limiar, causando um atraso na despolarização atrial e ventricular. A diminuição na fase 0 do potencial de ação leva a um potencial de ação maior, produzindo um atraso na condução intraventricular e atrioventricular. Na superfície do ECG, este irá apresentar um achatamento e perda das ondas P e alargamento dos complexos QRS. Com o crescente atraso na condução intraventricular, o ECG de superfície começa a apresentar sinais de bloqueio do feixe de ramos esquerdo e direito. Isto pode ser diferenciado da doença do feixe de ramos pelo fato de que na hipercalemia o atraso persiste em todo o complexo QRS, não apenas durante a porção inicial ou terminal, respectivamente.
- A 10 mEq/L, a condução sinoatrial não mais ocorre e o ritmo juncional acelerado toma conta. Arritmias ventriculares se desenvolvem com a fusão dos complexos QRS alargados com as ondas T para eventualmente formar o padrão clássico de onda sinusoidal. Uma vez que isto ocorre, a FV e assistolia são iminentes e a parada cardíaca irá então ocorrer.
- Algumas vezes as mudanças podem ser erráticas e imprevisíveis e o ECG saltará do normal para assistolia devido à variabilidade dos fatores etiológicos e seus efeitos influentes, por exemplo, taxa de mudança de potássio, concentração de cálcio, pH, e concentração de sódio. Assim, a hipercalemia deve ser tratada de forma emergente sempre que os níveis de potássio se tornarem maiores que 6,5 mmol/L, ou na presença de manifestações de ECG de hipercalemia, independentemente do nível de potássio. Outras associações relatadas com hipercalemia aguda incluem: quadro de pseudo IM no registro do ECG, com segmento ST-T maciço como resultado de desarranjos na repolarização de miócitos, pequenos intervalos PR e QT, taquicardia sinusal, bradicardia sinusal, ritmo idioventricular, bloqueio cardíaco de 1º e 2º graus.
Efeitos metabólicos
Hipercalemia leva à acidose metabólica hiperclorêmica, pois a hipercalemia promove a captação intracelular de potássio em troca de íons hidrogênio. Isto cria alcalose intracelular, suprimindo a produção de amônia renal nos túbulos proximais, levando a uma diminuição na excreção urinária de amônio e ácido e a uma acidose tubular renal tipo IV.
Bomba de potássio de sódio
A ATPase de sódio potássico foi descoberta em 1957 por Skou, que mais tarde recebeu uma parte do Prêmio Nobel de Química de 1997 por sua descoberta.
Skou foi o primeiro a descobrir a ATPase de sódio potássico no sarcolemma da superfície celular do músculo cardíaco. Sua presença foi posteriormente detectada em cada organismo eucariótico único e multicelular.
A bomba de potássio de sódio funciona ligando a hidrólise de ATP à exportação celular de três íons de sódio em troca de dois íons de potássio contra seus gradientes eletroquímicos. É o alvo molecular para digitalis e digoxina, que estão em uso desde o século 18 como extratos de dedaleira.
A ação da bomba de potássio sódico é regulada por um fosfoproteína fosfolimã, cuja não fosforilação leva à inibição da bomba e cuja fosforilação leva a um aumento da atividade da bomba. Possui três sítios de fosforilação, dois sítios de palmitelação e um sítio de glutationa, o que explica a multiplicidade de sinais capazes de estimular e inibir a bomba.
A própria bomba de potássio de sódio é uma enzima composta de múltiplas subunidades com múltiplas isoformas. A presença das subunidades alfa e beta (principalmente B1 no coração) é essencial para a sua função. Recentemente, uma terceira subunidade gama de proteínas foi identificada nos rins, mas até hoje sua função permanece desconhecida.
A subunidade alfa é o núcleo catalítico da enzima bomba de potássio de sódio. É aproximadamente 100 kDa e contém os locais de ligação de esteróides de sódio, potássio, ATP e cardiotônicos, como o ouabain. Apenas alfa 1 e alfa 2 apresentam uma presença significativa em um miócito cardíaco normal e estão funcionalmente ligados ao trocador de cálcio de sódio (NCX). Alpha 3 tem sido relatado para substituir alpha 2 em modelos experimentais de insuficiência cardíaca .
Dados de experimentos recentes favorecem o envolvimento de ambas subunidades alfa 1 alfa 2 da bomba na regulação do acoplamento excitação-contração (E-C). Pensa-se que o alfa 1, cuja expressão se encontra mais uniformemente distribuída pelo sarcolemma, desempenha um papel mais “doméstico”, controlando tanto a contratilidade como o sódio intracelular a granel, enquanto que o alfa 2 cuja expressão está concentrada nos tubos T juntamente com outros componentes chave do acoplamento E-C se pensa que se concentra principalmente na contratilidade .
Factores conhecidos que podem controlar a bomba de potássio de sódio incluem: ATP, sódio intracelular, barreiras sub-sarcolemmal e espaços difusos, potencial de membrana, vias de sinalização intracelular (vias de sinalização adrenérgica, proteína quinase A & C, óxido nítrico, fosfoliman), regulação direta por pequenas moléculas (lipídios, esteróides cardiotônicos endógenos), outras proteínas associadas (caveolae e caveolinas, e anquirina).
Conclusão
Hiperkalemia é um desafio clínico e pode se apresentar em até 10% dos pacientes hospitalizados. Seu resultado final é um risco de vida. Como todas as células do corpo são, em última instância, afetadas pela bomba de potássio sódico, e os músculos cardíacos isquêmicos são conhecidos por extrudar extracelularmente o potássio, levando a uma redução do limiar de arritmia com possibilidade de arritmias ventriculares que agravam a hipopolarização e diminuem ainda mais o limiar, mais estudos precisam ser focados na manipulação da enzima potássio sódico, pois seu controle poderia alterar favoravelmente os resultados das paradas cardíacas e reescrever as diretrizes atuais de RCP.