ABOVE: modificado de © ISTOCK.com, tera vector
Almost sempre, construir algo é mais difícil do que derrubá-lo. Da mesma forma, bater nos genes representa um desafio maior do que derrubá-los. É uma realidade que os pesquisadores terão que superar para tirar o máximo proveito da edição de genes. A colisão de genes permite aos cientistas estudar os efeitos de variantes específicas de genes, usar genes repórteres como a proteína verde fluorescente para rastrear produtos gênicos no tempo e no espaço, para sondar a regulação do genoma e, por fim, para reparar genes causadores de doenças. “É uma forma realmente eficaz de interrogar cada base de um gene”, diz Greg Findlay, um candidato a MD/PhD na Universidade de Washington.
CRISPR-Cas9, uma tecnologia de edição de genes conhecida por sua facilidade de uso, pode derrubar genes para dentro ou para fora. A eliminação de um gene envolve a inserção do CRISPR-Cas9 em uma célula usando um RNA guia que visa a ferramenta para o gene de interesse. Lá, o Cas9 corta o gene, cortando através de ambas as vertentes do DNA, e o mecanismo regular de reparação do DNA da célula fixa o corte usando um processo chamado de união final não-homológica (NHEJ). A NHEJ é altamente eficiente, mas imprecisa. O processo tende a introduzir erros na forma de pequenas inserções ou deleções que normalmente são suficientes para derrubar o gene.
Para derrubar um gene, entretanto, os cortes devem ser reparados com muita precisão, sem inserções ou deleções extras. Isto requer o aproveitamento de um segundo mecanismo de reparo do DNA chamado de reparo dirigido por homólogos (HDR), que – em células de mamíferos, pelo menos – ocorre de forma menos eficiente, de modo que sua freqüência é reduzida pela do NHEJ. Complicando ainda mais o processo está o fato de que alguns loci gênicos e tipos de células são inerentemente menos hospitaleiros para a edição do CRISPR-Cas9.
Nos últimos anos, os pesquisadores desenvolveram muitas novas estratégias para aumentar a eficiência de bater em genes grandes e pequenos usando o CRISPR-Cas9, e ao longo do caminho eles propuseram e testaram novas aplicações para este tipo de edição de genes. Aqui, The Scientist explora algumas das abordagens mais promissoras.
Select It
Researcher: Jon Chesnut, diretor sênior de biologia sintética R&D, Thermo Fisher Scientific
Projeto: Ao desenvolver um kit de marcação genética chamado Truetag que a Thermo Fisher colocará no mercado no final deste ano, Chesnut usou marcadores selecionáveis para melhorar
eficiência. Um marcador selecionável – neste caso, um gene de resistência a antibióticos – é colado a um tag protéico fluorescente e batido em
células mamíferas. Essas células são então cultivadas em cultura com o antibiótico associado. O gene de resistência confere uma vantagem seletiva às células que o carregam; só elas são capazes de crescer, e assim aquelas que crescem contêm o marcador genético de interesse. Mesmo que a eficiência de inserção do gene seja baixa, os pesquisadores podem usar a seleção antibiótica por uma semana ou mais para acabar com uma alta porcentagem de células com inserções bem sucedidas.
Usando a puroamicina ou blasticidina antibiótica com o kit, a equipe de Chesnut conseguiu aumentar a taxa de inserção do gene de 10-30 por cento para 90 por cento ou mais em algumas populações de células. Alguns genes especialmente difíceis passaram de uma taxa de inserção de menos de 1% para mais de 90%. É importante testar várias doses de antibióticos na linha celular que você planeja usar para encontrar a dose correta, Chesnut diz: você quer matar células sem inserções mas não células com inserções bem sucedidas.
Try It: Os marcadores seleccionáveis funcionam melhor quando o gene de interesse é altamente expresso, diz Chesnut. “Se não for, você ainda pode obter seleção, mas pode não obter expressão suficiente do seu marcador de proteína fluorescente para ser capaz de detectá-lo”. Também se aplicam as limitações gerais do CRISPR-Cas9. “Há regiões do genoma que não cortam muito bem com o CRISPR, e ainda não temos a certeza do porquê”, acrescenta. E alguns tipos de células não aceitam facilmente DNA estranho, RNA ou complexos RNA-proteína – os três métodos de entrega do CRISPR-Cas9.
Para melhor sorte na inserção de marcadores selecionáveis, certifique-se de que haja uma chamada seqüência PAM, uma pequena tag no DNA alvo que o CRISPR-Cas9 deve reconhecer antes de cortar, dentro de 10 pares de base do local desejado de inserção do gene, diz Chesnut. Mais longe do local de corte do que isso, e a eficiência da inserção pode ser muito baixa para ser funcional. Sem um site PAM, você pode tentar TALENs ou núcleos de dedos de zinco, embora aquelas técnicas de edição de genes mais antigas sejam mais complicadas que CRISPR.
Inibição Temporizada
Pesquisador: Jacob Corn, biólogo do genoma, Instituto Federal Suíço de Tecnologia, Zurique
Projeto: Os pesquisadores não entendem porque o caminho do NHEJ supera amplamente o caminho do HDR em células de mamíferos. “Levedura faz HDR como loucura”, diz Corn. Em um esforço para reverter esse processo de reparo do DNA em células humanas e melhorar o controle de knock-in dos genes, ele e sua equipe estão tentando identificar como o HDR é regulado. Eles examinaram células humanas em busca de genes cuja eliminação levou ao aumento da HDR na célula, e depois procuraram por pequenos inibidores de moléculas desses genes. Um dos genes que apareceu nos códigos do CDC7, uma cinase que regula a transição do ciclo celular para a fase S; seu inibidor, XL413, aumentou a eficiência de knock-in do gene duas ou três vezes (BioRXiv, DOI: 10.1101/500462, 2018). Isso porque o HDR ocorre apenas em algumas partes do ciclo celular, incluindo a fase S, diz Corn. Se você adicionar o inibidor XL413 ao mesmo tempo que usa o CRISPR-Cas9 para editar seu gene alvo, as células se acumulam na fase imediatamente anterior à fase S. Então você remove o XL413, e todas as células entram na fase S e aumentam a eficiência de knock-in.

Corn tem usado esta técnica em muitas linhas de células humanas imortalizadas e em células T humanas. Ela pode bater em pequenos trechos de DNA, tais como SNPs, bem como em grandes genes. Não há razão para não funcionar em ratos, diz ele, embora não a tenha testado.
Try It: “O tempo é absolutamente fundamental”, diz Corn. O Cas9 deve cortar o DNA ao mesmo tempo em que o XL413 é adicionado. Se você inibir primeiro e depois liberar enquanto edita com o CRISPR-Cas9, a eficiência da recombinação homóloga cai três vezes ao invés de aumentar, porque as células são liberadas na fase errada do ciclo celular.
E como em qualquer esforço HDR, Corn diz, sempre execute um controle sem liberação para ter certeza de que você não está amplificando acidentalmente o DNA contaminante que está flutuando ao redor do seu laboratório. Depois de introduzir o knock-in, “seqüência, seqüência, seqüência, seqüência”, diz ele. Só usando um sistema de repórter, como uma etiqueta de proteína fluorescente, para demonstrar a inserção bem sucedida do gene pode ter um tiro pela culatra. A sequência verifica que as inserções foram feitas no local correto.
Jogando o Jogo Longo
Pesquisador: Channabasavaiah Gurumurthy, director do núcleo de engenharia do genoma do rato, Centro Médico da Universidade de Nebraska
Projecto: Há alguns anos atrás, pensando sobre a dificuldade de bater em genes enquanto tentava fazê-lo em zigotos de rato, Gurumurthy e seus colegas tiveram uma revelação.
Pesquisadores estavam inserindo com sucesso DNA curto, de cadeia única, então por que não tentar fazer um knock-in inserindo DNA longo, de cadeia única? De fato, a abordagem, que Gurumurthy chama Easi-CRISPR (adições eficientes com inserções de ssDNA -CRISPR), aumenta a eficiência em 2,5 vezes, e usando DNA de uma única corda corta a taxa de inserções fora do alvo 100 vezes na cultura de células (Nat Protoc 13:195-215, 2018; Nature 559:405-09, 2018). “É bastante grande”, diz ele. No laboratório de Gurumurthy, Easi-CRISPR gerou uma linha de rato em knock-in para 9 em cada 10 genes que eles tentaram. Um colaborador também a usou em células T humanas para criar células CAR-T, células imunes específicas do paciente para combater o câncer.
Try It: Easi-CRISPR está longe de ser infalível, Gurumurthy cautelas. Às vezes a técnica insere apenas parte do gene. Além disso, ele acrescenta, ela pode embaralhar os braços da homologia – sequências curtas em ambos os lados do gene que o aloja ao seu alvo correto no genoma. E alguns loci são inexplicavelmente mais difíceis de inserir do que outros.
Poucos vendedores comerciais projetam e sintetizam DNA personalizado longo e de cadeia única. Você pode fazer o seu próprio, mas a estabilidade do DNA de cadeia única varia; seqüências menos estáveis terão rendimentos menores, então você pode precisar sintetizar mais deles, diz Gurumurthy.
Pesquisadores incapazes de inserir CRISPR em embriões de camundongo de célula única podem pagar uma facilidade central para fazer os ratos com sua seqüência de DNA, diz Gurumurthy. Instalações centrais como a sua cobrança de $5,000-$15,000 para gerar um ou dois pares de reprodução; instalações comerciais cobram $20,000-$50,000, diz ele.
Knock-in By Numbers
Pesquisador: Greg Findlay, candidato a MD/PhD no laboratório de Jay Shendure, Universidade de Washington
Projeto: Findlay e seus colegas tinham como objectivo melhorar a forma como os clínicos interpretam as mutações no gene BRCA1 do cancro da mama e dos ovários. Esse gene tem milhares de variantes, mas os pesquisadores não sabem como a maioria delas afeta sua função. Para estudar o impacto dessas variantes, eles utilizaram uma técnica de knock-in que desenvolveram chamada edição do genoma de saturação (Nature, 562:217-22, 2018).
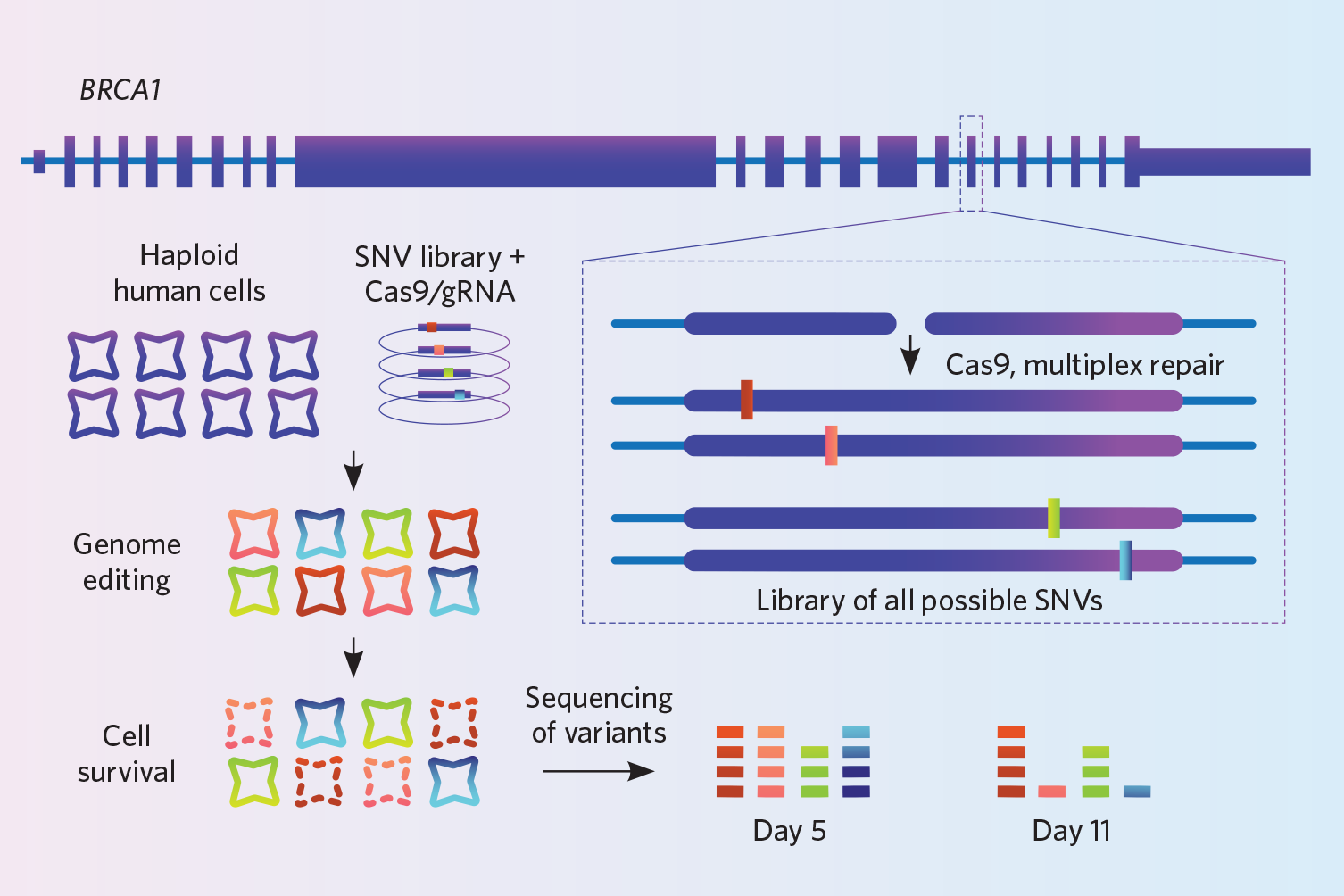
Em uma linha imortalizada de células humanas haplóides, eles usaram CRISPR-Cas9 para bater em 4.000 pequenas variantes em milhões de células de uma vez in vitro. O genoma é cortado no mesmo local em cada célula, mas o genoma de cada célula recebe uma variante diferente. Para promover a HDR, eles também eliminaram o gene ligase4, desativando o caminho de reparo da NHEJ – um passo que resultou em um triplo ganho de eficiência, diz Findlay. Finalmente, como todos os knock-ins das células são diferentes, eles sequenciaram as células profundamente, cobrindo a mesma região genômica milhões de vezes, para ter certeza de que elas realmente batiam nas 4.000 variantes que eles queriam estudar. Eles se sequenciaram em dois pontos de tempo, e deduziram que os knock-ins que não surgiram na sequência no segundo ponto de tempo foram aqueles que interferiram com a função do gene, porque as células que as carregavam devem ter morrido.
Try It: A equipa da Findlay mandou fabricar os oligos de ADN para as 4.000 variantes num microarray. Você pode comprar matrizes de 6.000 a 250.000 oligo-elementos, então pense em obter mais “bang” pelo seu dólar combinando várias experiências na mesma matriz, diz Findlay. O laboratório deles paga cerca de $5.000 por 100.000 oligo-elementos.
Esta estratégia vem com limitações: até agora só foi usada para bater em variantes de um único nucleotídeo, e todas as edições precisam estar no mesmo gene. O método funciona melhor quando se edita uma região bastante estreita de DNA, cerca de 110-120 pares de bases, porque os oligoetideos de DNA mais longos teriam muitos erros, diz Findlay. Também é importante fazer uma sequência muito profunda para garantir que você contabilize o número total de variantes que você pretendia introduzir.