- A Revisão Clinicopatológica
- INTRODUÇÃO
- EPITHELIAL-STROMAL CORNEAL DYSTROPHIES
- DISTROFIA DE CORNEAL DE IRIS-BUCKLERS
- Distrofia corneana
- DISTROFIA CORNEAL GRANULAR, TIPO I
- DISTROFIA CORNEAL GRANULAR, TIPO II
- DISTROFIAS CORNEALESTROMAIS
- DISTROFIA CORNEAL MACULAR
- DISTROFIA CORNEAL DE SCHNYDER (SCD)
- Tabela 4: Mnemónica para recordar as distrofias do estroma da córnea
- OVERVIEW: DISTROFIA ESTROMAL CORNEAL
- EPIDEMIOLOGIA
- SIGNS
- SYMPTOMS
- TREATAMENTO
A Revisão Clinicopatológica
Emily S. Birkholz, MD, Nasreen A. Syed, MD, e Michael D. Wagoner, MD, PhD
Agosto 17, 2009
Revisão Maior: Chaunhi Van, MD e Nasreen Syed, MD
Agosto 20, 2015
INTRODUÇÃO
Distrofias epiteliais e estromais corporais são um grupo de distrofias hereditárias da córnea que são causadas pelo acúmulo progressivo de depósitos dentro das camadas da córnea. Estes depósitos não são causados por inflamação, infecção ou trauma, mas por mutações genéticas que levam à transcrição de proteínas aberrantes resultando no acúmulo de material insolúvel dentro da córnea. Os distúrbios podem ou não afetar a visão e podem ou não ser simétricos (1). O sistema de classificação do Comité Internacional para a Classificação das Distrofias da Córnea de 2015 (IC3D) dividiu as distrofias da córnea em 4 categorias: distrofias epiteliais e subepiteliais, distrofias epiteliais-estromais, distrofias estromais, distrofias estromais e distrofias endoteliais. A maioria das distrofias anteriormente consideradas distrofias estromais são agora classificadas como distrofias epiteliais-estromais ou distrofias estromais. As tabelas 1 e 2 listam as distrofias epiteliais-estromais e as distrofias estromais (2). A antiga classificação para as distrofias do estroma da córnea está listada na Tabela 3.
- Distrofia corneana Reis-Bucklers
- Distrofia corneana Thiel-Behnke
- Distrofia corneana em malha, tipo 1 e variantes
- Distrofia da córnea granular, tipo 1
- Distrofia da córnea granular, tipo 2
- Distrofia da córnea macular
- Distrofia da córnea Schnyder
- Distrofia da córnea estromal congénita
- Distrofia da córnea amorfa posterior
- Distrofia central da córnea nublada de François
- Distrofia pré-Descemet corneal dystrophy
Tabela 3. Antiga classificação de distrofias estromais da córnea
- Distrofia da córnea da camada de estrofe
- Distrofia da córnea granular
- Distrofia da córnea de Avellino
- Distrofia da córnea macular
- Gotas gelatinosascomo a distrofia
- Distrofia corneana de Schnyder
- François-Neetans Fleck dystrophy
- Distrofia hereditária congênita do estroma
>
>
EPITHELIAL-STROMAL CORNEAL DYSTROPHIES
Distrofias estromais epiteliais são causadas por mutações no gene do fator de crescimento transformador beta-induzido (TGFβI), também conhecido como o gene BIGH3. TGFβI está localizado no cromossoma 5q31 e códigos para queratoepithelin, uma proteína secretada pelo epitélio corneano. Esta proteína atua como uma proteína de adesão e está presente no estroma normal. Sendo uma proteína pequena aproximadamente do tamanho da albumina, ela tem a capacidade de se difundir através do estroma corneano. Quando ocorre uma mutação no gene TGFβI, a estrutura da queratoepitelina é anormal e o acúmulo da proteína insolúvel ou seus fragmentos proteolíticos ocorre na córnea (1, 3). Curiosamente, a mutação do gene TGFβI foi descoberta em parte na Universidade de Iowa. Um grupo de pesquisadores e clínicos incluindo Edwin M. Stone, Robert Folberg e Jay H. Krachmer mapeou granular tipo I, granular tipo II, e distrofia da malha ao cromossomo 5q em 1994 (4). Até o momento, 63 mutações diferentes foram identificadas no gene TGFβI. Não foram identificados tratamentos eficazes para prevenir ou atenuar a deposição do queratoepithelin. As distrofias têm tipicamente uma herança autossômica dominante e envolvem a camada Bowman e o estroma (3).
DISTROFIA DE CORNEAL DE IRIS-BUCKLERS
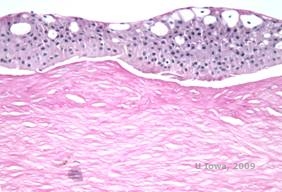
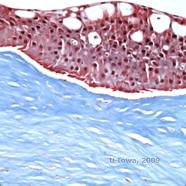
Reis-Bücklers, anteriormente conhecida como Distrofia da Córnea Granular tipo III ou Distrofia da Córnea de Bowman tipo I, tipicamente presente com córneas normais ao nascimento mas desenvolvendo erosões recorrentes dolorosas, opacificação, e perda progressiva da visão na primeira década de vida (1). Opacidades irregulares, cinzento-branco, de tipo geográfico, estão localizadas na camada Bowman e estroma anterior. Em estágios mais avançados da doença, as opacidades podem se estender até o limbo e estroma mais profundo (2). A histopatologia revela depósitos anteriores de estroma e subepiteliais de material hialino que perturbam e frequentemente substituem a camada de Bowman (ver Figura 1A e 1B). Os depósitos mancham de vermelho com a coloração de tricromio de Masson (2). O material semelhante à hialina consiste em corpos semelhantes a varas por ultra-estrutura, o que ajuda a distingui-lo da distrofia corneana de Thiel-Behnke (1, 2).
| A: H&E de Reis Bückler mostrando destruição da camada de Bowman e epitélio irregular | B. Coloração tricromada de Masson demonstrando coloração epitelial |
 |
 |
Distrofia corneana
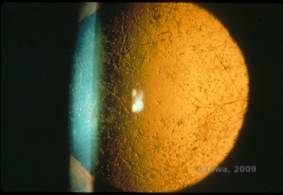
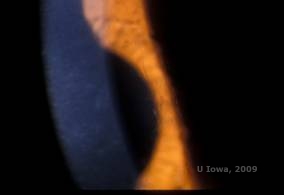
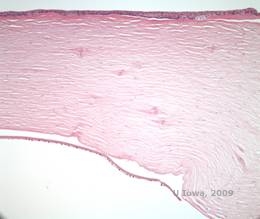
Distrofia corneana (LCD) é a mais comum das distrofias epiteliais estromais corneanas. É tipicamente uma doença autossômica dominante, bilateral, que tipicamente se apresenta no final da primeira década de vida com sintomas de erosões recorrentes da córnea e diminuição da visão. Caracteriza-se por linhas de treliça que são lineares, orientadas radialmente, ramificando opacidades refratárias descritas como “tipo vidro”, localizadas no estroma anterior (ver Figura 2A e 2B). Estas linhas de treliça são inicialmente encontradas na córnea central superficial. À medida que a doença progride, elas se espalham de forma mais profunda e periférica no estroma, com pouquíssimo esforço do limbo (1, 2). Outros achados de exame incluem opacidades tipo pulga, pontos brancos subepiteliais e névoa de estroma “ground-glass”, que começa centralmente e se torna mais difusa (2). Muitos pacientes com LCD necessitarão de intervenção cirúrgica para tratar erosões recorrentes e diminuição da visão. Se a doença estiver localizada anteriormente no estroma, os pacientes podem muitas vezes ser tratados com sucesso com keratectomia fototerapeutica (PTK). Alguns requerem transplante de córnea. Como a queratoepitelina, a proteína produzida pelo gene TGFβI, é produzida principalmente no epitélio corneano, a doença tende a recidivar nos enxertos corneanos (1).
Em LCD, depósitos amilóides acumulam-se entre a membrana epitelial do porão e a camada Bowman, assim como no estroma, causando distorção da arquitetura lamelar. Os depósitos mancham positivamente com imunohistoquímica usando anticorpos contra queratoepithelin (2). Os depósitos aparecem como depósitos amorfos rosa sobre a hematoxilina e eosina (H&E) (ver figura 1C e 1D) e coloração com vermelho Congo demonstrando a clássica birefringência verde maçã na polimerização cruzada (ver figura 2E e 2F) (1). Ausência ou desbaste da camada Bowman, atrofia epitelial e degeneração epitelial basal também podem ser encontradas na histopatologia em LCD (2).
LCD tipo I é a forma clássica de LCD causada por uma mutação no gene TGFβI resultando em deposição amilóide isolada na córnea. Quatro variantes do LCD foram identificadas: LCD tipo IIIA, tipo I/IIIA, tipo IV, e amiloidose polimórfica. Variantes do LCD presentes mais tarde que o LCD clássico. O LCD tipo IIIA apresenta-se na 5ª-7ª década, geralmente com erosões epiteliais. Possui linhas de malha mais grossas, descritas como “ropy-appearing”, que se estende até o limbo. O LCD tipo I/IIIA tem linhas de malha fina. O LCD tipo IV apresenta-se na 7ª-9ª década com pequenas linhas de treliça. Depósitos amilóides no LCD tipo IV são encontrados no estroma profundo e raramente ocorrem erosões epiteliais. As linhas de treliça estão ausentes na amiloidose polimórfica e raramente ocorrem erosões epiteliais (2).
LCD tipo II é uma síndrome de amiloidose sistêmica conhecida como síndrome de Meretoja afetando a pele, nervos cranianos e córnea. Apresenta-se no início da idade adulta com neuropatias periféricas, neuropatias cranianas, fácies tipo cão de caça, pele seca, blefarocalasia, lábios salientes e linhas de treliça corneana. Este tipo foi ligado ao gene da gelsolina no cromossomo 9, que codifica uma proteína precursora amilóide que funciona para remover a actina dos locais de lesão e inflamação (1). O nome é um erro de nome e não é considerado uma variante da distrofia da córnea da malha (2).
| A: Olho esquerdo na retroiluminação demonstrando depósitos estromais anteriores na distrofia corneana da malha | B: Olho esquerdo com maior potência mostrando depósitos estromais anteriores lineares. |
 |
 |
| C: H&E: Coloração da córnea com trama. Observar depósitos amorfos rosa no estroma | D: Uma visão mais próxima dos depósitos amorfos rosa |
|
|
| E: Corante vermelho Congo, realçando amilóide | F: Birefringência verde maçã de amilóide com polarização cruzada. |
 |
 |
DISTROFIA CORNEAL GRANULAR, TIPO I
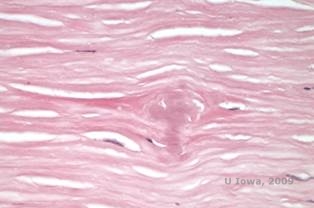
Distrofia corneana craniana, tipo I (GCD1) é uma doença bilateral, autossômica dominante associada a uma mutação no gene TGFβI que leva à deposição de um material hialino no estroma corneano. Apresenta-se tipicamente no início da primeira década de vida com opacidades cinzento-branco, “tipo migalhas” no estroma anterior a médio, estendendo-se para o estroma posterior na doença do avanço (1, 2). Essas opacidades são depósitos discretos localizados centralmente, com córnea clara localizada na periferia e córnea clara entre depósitos (ver Figura 3A e 3B). A doença é tipicamente assintomática no início, mas com o tempo as opacidades podem coalescer e levar à diminuição da visão. Erosões recorrentes da córnea podem ocorrer na DCG, mas com uma incidência menor do que na DCL (1, 5). Os pacientes também podem experimentar ofuscamento e fotofobia (2). O tratamento no início do processo da doença é muitas vezes apenas observação. Entretanto, conforme a doença progride, o PTK e o transplante de córnea podem ser necessários para melhorar os sintomas de visão e erosão. Tal como o LCD, a doença pode ocorrer em enxertos corneanos.
Histopatologicamente, as opacidades são depósitos eosinofílicos muitas vezes descritos como “tipo rocha doce” no estroma anterior feito de um material parecido com hialina. Com o tempo, os depósitos progridem para o estroma mais profundo da córnea. O material hialino mancha de vermelho vivo com coloração tricromada Masson (Veja Figura 3C e 3D).
| A:Foto de lâmpada de fenda da Distrofia da Córnea Granular, tipo I | B: Note os depósitos de estroma “crumb-like” com estroma interveniente claro. |
 |
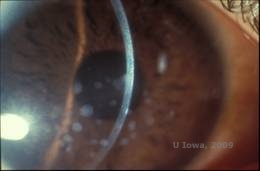 |
| C: H& E mancha de córnea mostrando depósitos hialinos eosinófilos “tipo rocha doce” em estroma | D: Coloração de material hialino vermelho vivo com Masson-Trichrome |
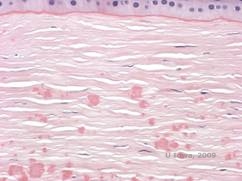 |
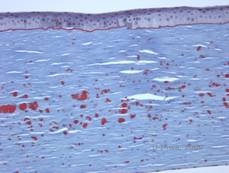 |
DISTROFIA CORNEAL GRANULAR, TIPO II
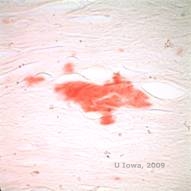
Distrofia corneana craniana, tipo II (GCD2), anteriormente conhecida como Avellino ou distrofia corneana combinada granulo-latídica, é uma doença autossômica dominante ligada a uma mutação no gene TGFβI que leva a um depósito tanto de hialina quanto de amilóide no estroma corneano. Tipicamente, pacientes presentes na segunda década de vida com pequenos pontos brancos-acinzentados no estroma superficial. As opacidades também podem aparecer em forma de espinhos, anel ou estelato. Na retroiluminação, eles são parcialmente translúcidos. Mais tarde, no processo da doença, podem também desenvolver linhas de grelha (ver figura 4A e 4B). Estas linhas não se cruzam e parecem mais brancas e menos refractárias do que as linhas da malha. Os sintomas do GCD2 são dor com erosões epiteliais e deficiência visual (2).
Histopatologicamente, a córnea terá depósitos estromais que mancham de vermelho com Tricromio de Masson, indicando a presença de hialina (Ver Figura 4C). Além disso, a coloração com vermelho Congo demonstrará birefringência verde maçã na polimerização cruzada indicando a presença de amilóide (Ver Figura 4D). Pensava-se que a doença era originária de uma família em Avellino, Itália. Entretanto, GCD tipo II tem agora sido relatado em pacientes de muitos outros países também (2,5), sendo a maior prevalência no leste da Ásia.
| A: Distrofia de Avellino mostrando depósitos semelhantes à malha e depósitos semelhantes à granular no estroma corneano | B. Coloração tricromada de Masson demonstrando depósitos de hialina estromal anterior |
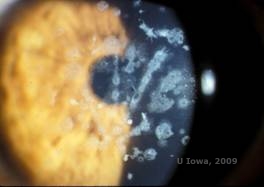 |
|
| C: Coloração vermelha do Congo mostrando depósitos de amilóide amorfo e rosa no mesmo espécime corneano. | D: A polimerização cruzada revela birefringência verde maçã, indicando amilóide. |
 |
 |
DISTROFIAS CORNEALESTROMAIS
DISTROFIA CORNEAL MACULAR
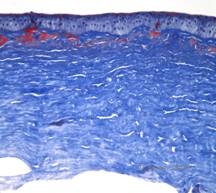
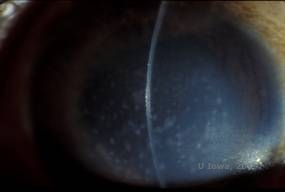
Distrofia da córnea macular (MCD) é uma doença autossômica recessiva causada por uma mutação no gene da sulfo-transferase 6 de carboidratos (CHST6) no cromossomo 16 que leva a um defeito na síntese do sulfato de queratana, o glicosaminoglicano principal da córnea. É o menos comum do que o LCD ou GCD, mas tende a ter um impacto mais severo na visão. Embora o DCM seja menos comum no mundo inteiro que o LCD ou DCG, é a mais comum das distrofias estromais da córnea em lugares como Islândia e Arábia Saudita (2,6). Lesões estromais anteriores semelhantes ao GCD1 aparecem na córnea na primeira década de vida, de cor cinza-esbranquiçada e em forma de pulga. Ao contrário do GCD1, no entanto, existe uma neblina estromal entre os depósitos e toda a córnea, de limbo a limbo está frequentemente envolvida (ver Figura 5A e 5B). A córnea é fina e à medida que a doença progride a membrana Descemet torna-se cinzenta e desenvolve as tripas. Erosões epiteliais podem ocorrer, mas menos no DMC do que no LCD. Os pacientes normalmente desenvolvem perda visual severa na segunda a terceira década de vida devido à névoa córnea difusa. O PTK pode ser realizado em alguns casos iniciais de CDM. No entanto, esta condição geralmente não é tão favorável ao PTK quanto a malha ou distrofia granular e freqüentemente requer transplante da córnea para tratamento (7). A recorrência em enxertos é menos comum no CDM do que na distrofia granular ou da malha (1,2,5,6,8).
Os depósitos estromais no CDM são compostos de mucopolissacarídeos que se acumulam dentro do retículo endoplasmático de queratócitos do estroma corneano, extracelularmente entre as lamelas estromais, e dentro do epitélio, membrana Descemet e endotélio. Estes depósitos apresentam coloração azul com azul de Alcian (ver figura 5C e 5D) (1). Há quebras na camada Bowman e intestino com espessamento da membrana Descemet (2).
Três subtipos de DMC foram descritos com base na presença ou ausência de sulfato de queratana imunoreativa em vários tecidos. O tipo I não possui sulfato de queratina imunoreativa no estroma corneano, queratócitos, soros ou cartilagem, e é a variante mais comum do CDM em todo o mundo. O tipo IA não possui sulfato de queratina no estroma, soros e cartilagem, mas tem níveis detectáveis dentro dos queratócitos. O tipo II tem sulfato de queratina presente em níveis muito reduzidos no estroma, queratócitos, soros e cartilagem (6).
| A: Fenda da foto da distrofia da córnea macular. | B: Note a bruma entre os depósitos estromais da córnea |
 |
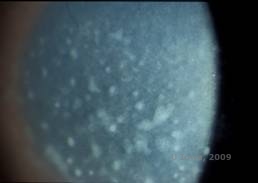 |
| C: H&E de córnea com distrofia macular. Observar depósitos estromais anteriores e ruptura da camada do Bowman | D: Depósitos de mucopolissacarídeos dentro de queratócitos realçados com coloração Alcian Blue |
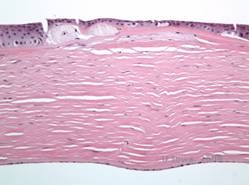 |
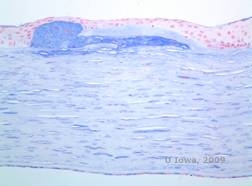 |
DISTROFIA CORNEAL DE SCHNYDER (SCD)
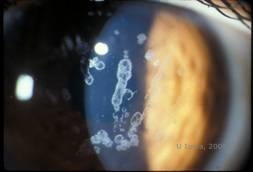
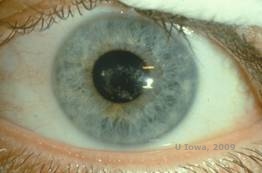
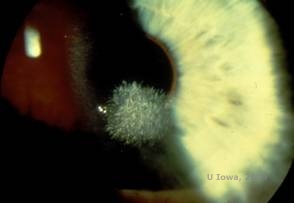
Distrofia corneana de Schnyder (SCD), anteriormente conhecida como distrofia cristalina da córnea de Schnyder, é uma distrofia autossômica dominante e bilateral da córnea ligada a uma mutação genética no domínio da preniltransferase UbiA contendo o gene 1(UBIAD1) no cromossomo 1. O defeito metabólico resultante dos queratócitos corneanos leva à deposição de colesterol cristalino no estroma. No entanto, a presença de cristais não é absolutamente necessária para o diagnóstico de SCD. Na verdade, apenas 54% dos pacientes com SCD possuem cristais de córnea. Tipicamente, os pacientes apresentam na segunda ou terceira década uma opacidade central da córnea em forma de anel com ou sem cristais subepiteliais em forma de vírgula (ver Figura 6A e 6B). Em seguida, lipoides arcus aparecem entre os 23 e 38 anos de idade. Após os 38 anos de idade, a turvação progressiva da córnea resulta numa neblina panstromal que atinge a medioperiferia. A maioria dos pacientes acima dos 50 anos de idade tem perda de visão fotópica, ofuscamento e diminuição da sensação corneana e, portanto, pode requerer tratamento cirúrgico, incluindo transplante de córnea ou PTK. Pode ocorrer recidiva no enxerto. A doença tem sido associada com hipercolesterolemia, hiperlipidemia e verdadeira valgum em alguns pacientes (2,5,9,10).
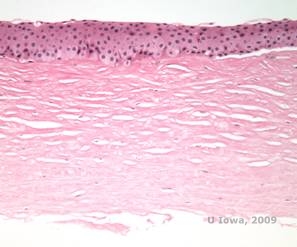
Histopatologicamente, cristais de colesterol birefringentes compostos de fosfolipídios e depósito de colesterol dentro das células epiteliais basais, queratócitos, camada de Bowman e entre as lamelas do estroma. Os lipídios dissolvem-se no processamento histológico normal, portanto cortes congelados através da córnea devem ser obtidos para demonstrar a presença de lipídios com Oil-Red-O ou manchas negras do Sudão.
| A: Fotografia de Schnyder Corneal Distrophy. | B. Depósitos cristalinos localizados centralmente |
 |
 |
| C: H&E de córnea com SCCD | D. A mancha Oil Red O destaca os cristais de colesterol que parecem vermelhos. |
 |
 |
Tabela 4 fornece uma mnemónica comum para memorizar algumas das distrofias da córnea que afectam o estroma, a composição do seu depósito, e o método de coloração destes depósitos é listado.
Tabela 4: Mnemónica para recordar as distrofias do estroma da córnea
- Distrofia Marilyn-Macular
- Monroe-Mucopolissacarídeo
- Coloração sempre azul alciano
- Gets-Granular Distrofia
- Her-Hialina
- Man in-Masson Trichrome stain
- Los-Lattice Dystrophy
- Angeles-Amyloid
- California-Congo Red
OVERVIEW: DISTROFIA ESTROMAL CORNEAL
EPIDEMIOLOGIA
|
SIGNS
|
SYMPTOMS
|
TREATAMENTO
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Patologia Conjuntival e Corneal. Albert e Jakobiec’s Principles and Practice of Ophthalmology, 3ª edição. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D classification of corneal dystrophies–edição 2. Córnea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. Aspectos clínicos e genéticos das distrofias da córnea associadas ao TGFBI. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Três distrofias corneanas autossômicas dominantes mapeadas para o cromossomo 5q. Genet da natureza. 1994; 6: 47-51.
- Bron AJ. Genética das Distrofias da Córnea: O que aprendemos nos últimos vinte e cinco anos. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Queratoplastia Penetrante para Distrofia da Córnea Macular. Oftalmologia 2005;112: 220-224.
- Badr IA, Wagoner MD. Keratectomia Fototerapêutica para a Distrofia da Córnea Macular. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetics of anterior and stromal corneal dystrophies. Seminários em Oftalmologia, 2008; 23: 1,9-17.
- Weiss JS. Distrofia da córnea de Schnyder. Curr Opin Opinião Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. O gene da distrofia cristalina da córnea de Schnyder mapeia o cromossomo humano 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Formato de citação: Van C, Syed NA. Distrofias Epiteliais-Estromal e Corneal Estromal: Uma Revisão Clínico-opatológica. Revisão de ; EyeRounds.org. 20 de agosto de 2015. Disponível em: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm