- A Clinicopathologic Review
- INTRODUCTION
- EPITHELIAL-STROMAL CORNEAL DYSTROPHIES
- REIS-BUCKLERS CORNEAL DYSTROPHY
- LATTICE CORNEAL DYSTROPHY
- GRANULAIRE CORNEALE DYSTROPHY, TYPE I
- GRANULAIRE CORNEALE DYSTROPHY, TYPE II
- STROMALE CORNEALE DYSTROPHIEEN
- MACULAIRE CORNEALE DYSTROPHIE
- SCHNYDER CORNEAL DYSTROPHY (SCD)
- Tabel 4: Mnemonogram voor het onthouden van hoornvliesstromale dystrofieën
- OVERZICHT: CORNEALE STROMALE DYSTROPHIEEN
- EPIDEMIOLOGIE
- SIGNS
- SYMPTOMEN
- TREATMENT
A Clinicopathologic Review
Emily S. Birkholz, MD, Nasreen A. Syed, MD, and Michael D. Wagoner, MD, PhD
August 17, 2009
Major Revision: Chaunhi Van, MD and Nasreen Syed, MD
August 20, 2015
INTRODUCTION
Corneale epitheliale-stromale en stromale dystrofieën zijn een groep van erfelijke aandoeningen van het hoornvlies die worden veroorzaakt door progressieve ophoping van afzettingen binnen de lagen van het hoornvlies. Deze afzettingen worden niet veroorzaakt door ontsteking, infectie of trauma, maar door genetische mutaties die leiden tot de transcriptie van afwijkende eiwitten, wat resulteert in de ophoping van onoplosbaar materiaal binnen het hoornvlies. De aandoeningen kunnen al dan niet het gezichtsvermogen aantasten en kunnen al dan niet symmetrisch zijn (1). Het classificatiesysteem 2015 van het Internationaal Comité voor de classificatie van corneale dystrofieën (IC3D) heeft corneale dystrofieën ingedeeld in 4 categorieën: epitheliale en subepitheliale dystrofieën, epitheliale-stromale dystrofieën, stromale dystrofieën, en endotheliale dystrofieën. De meeste dystrofieën die vroeger als stromale dystrofieën werden beschouwd, worden nu ingedeeld als hetzij epitheliaal-stromale dystrofieën, hetzij stromale dystrofieën. Tabel 1 en 2 geven een overzicht van de epitheliale-stromale dystrofieën en stromale dystrofieën (2). De oude classificatie voor hoornvliesstromale dystrofieën is opgenomen in tabel 3.
- Reis-Bucklers corneal dystrophy
- Thiel-Behnke corneal dystrophy
- Lattice corneal dystrophy, type 1 en varianten
- Granulaire hoornvliesdystrofie, type 1
- Granulaire hoornvliesdystrofie, type 2
- Maculaire corneale dystrofie
- Schnyder corneale dystrofie
- Congenitale stromale corneale dystrofie
- Posterieure amorfe corneale dystrofie
- Centrale troebele dystrofie van Francois
- Pre-Descemet corneale dystrofie
Tabel 3. Oude classificatie van corneale stromale dystrofieën
- Lattice corneale dystrofie
- Granulaire corneale dystrofie
- Avellino corneale dystrofie
- Maculaire corneale dystrofie
- Gelatineachtige druppel-zoals dystrofie
- Schnyder hoornvliesdystrofie
- Francois-Neetans Fleck-dystrofie
- Congenitale erfelijke stromale dystrofie
EPITHELIAL-STROMAL CORNEAL DYSTROPHIES
Epithelial-stromal dystrophies worden veroorzaakt door mutaties in het transforming growth factor beta-induced (TGFβI)-gen, ook bekend als het BIGH3-gen. TGFβI bevindt zich op chromosoom 5q31 en codeert voor keratoepitheline, een eiwit dat door hoornvliesepitheel wordt afgescheiden. Dit eiwit fungeert als een adhesie-eiwit en is aanwezig in normaal stroma. Omdat het een klein eiwit is, ongeveer zo groot als albumine, heeft het de mogelijkheid om door het hoornvliesstroma te diffunderen. Wanneer een mutatie in het TGFβI gen optreedt, is de keratoepitheline structuur abnormaal en accumulatie van het onoplosbare eiwit of zijn proteolytische fragmenten treedt op in de cornea (1, 3). Interessant is dat de TGFβI genmutatie gedeeltelijk werd ontdekt aan de Universiteit van Iowa. Een groep onderzoekers en clinici, waaronder Edwin M. Stone, Robert Folberg, en Jay H. Krachmer brachten granulaire type I, granulaire type II, en rasterdystrofie in kaart op chromosoom 5q in 1994 (4). Tot op heden zijn er 63 verschillende mutaties in het TGFβI gen geïdentificeerd. Er zijn nog geen effectieve behandelingen om de afzetting van keratoepitheline te voorkomen of af te zwakken. De dystrofieën hebben meestal een autosomaal dominante overerving en betreffen de Bowman laag en stroma (3).
REIS-BUCKLERS CORNEAL DYSTROPHY
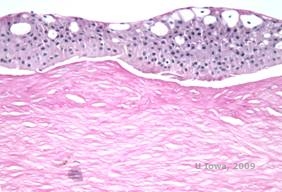
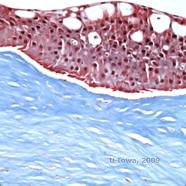
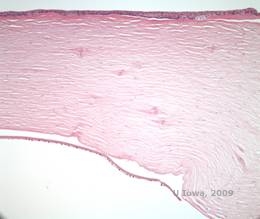
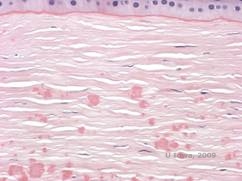
Reis-Bücklers, vroeger bekend als Granulaire hoornvliesdystrofie type III of Corneale Dystrofie van Bowman’s type I, vertonen bij de geboorte meestal normale hoornvliezen, maar ontwikkelen pijnlijke recurrente erosies, opacificatie en progressief gezichtsverlies in de eerste tien jaar van het leven (1). Onregelmatige, grijs-witte, geografisch-achtige opaciteiten zijn gelokaliseerd in de Bowman laag en de anterieure stroma. In verder gevorderde stadia van de ziekte kunnen de troebelingen zich uitbreiden tot de limbus en de diepere stroma (2). Histopathologie onthult anterieure stromale en subepitheliale afzettingen van hyalienachtig materiaal die de laag van Bowman onderbreken en vaak vervangen (zie figuur 1A en 1B). De afzettingen kleuren rood met Masson trichroomkleuring (2). Het hyaline-achtig materiaal bestaat uit staafvormige lichaampjes ultrastructureel, wat helpt om het te onderscheiden van Thiel-Behnke hoornvliesdystrofie (1, 2).
| A: H&E van Reis Bückler toont vernietiging van Bowman’s laag en onregelmatig epitheel | B. Masson Trichrome vlek die epitheliale kleuring toont |
 |
 |
LATTICE CORNEAL DYSTROPHY
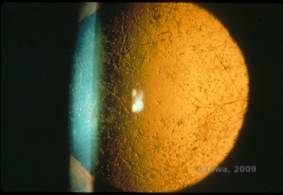
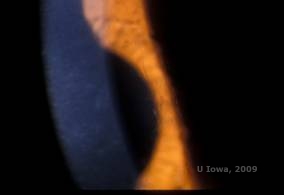
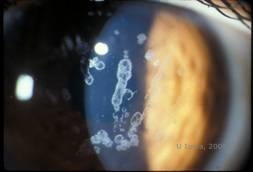
Lattice corneal dystrophy (LCD) is de meest voorkomende van de corneale epitheliale-stromale dystrofieën. Het is typisch een autosomaal dominante, bilaterale ziekte die zich typisch tegen het einde van het eerste decennium van het leven presenteert met symptomen van recurrente cornea-erosies en verminderd gezichtsvermogen. Ze wordt gekenmerkt door tralielijnen, lineaire, radiaal georiënteerde, vertakkende refractiele troebelingen, beschreven als “glasachtig” in het voorste stroma (zie figuur 2A en 2B). Deze rasterlijnen worden aanvankelijk aangetroffen in de oppervlakkige centrale cornea. Naarmate de ziekte vordert, verspreiden zij zich dieper en perifeer in het stroma met sparing van de limbus (1, 2). Andere onderzoeksbevindingen zijn vlekachtige troebelingen, subepitheliale witte stippen en “matglazen” stromale waas, die centraal begint en meer diffuus wordt (2). Veel patiënten met LCD hebben een chirurgische ingreep nodig voor de behandeling van terugkerende erosies en verminderd gezichtsvermogen. Als de ziekte zich anterieur in het stroma bevindt, kunnen patiënten vaak succesvol worden behandeld met fototherapeutische keratectomie (PTK). Sommigen hebben een hoornvliestransplantatie nodig. Omdat keratoepitheline, het eiwit dat geproduceerd wordt door het TGFβI gen, voornamelijk geproduceerd wordt in het hoornvliesepitheel, heeft de ziekte de neiging om terug te keren in hoornvliestransplantaties (1).
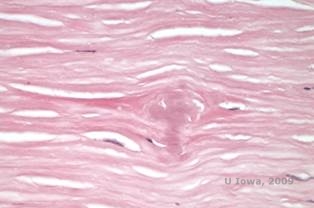
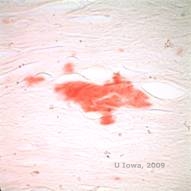
Bij LCD hopen amyloïde afzettingen zich op tussen het epitheliale basaalmembraan en de Bowman laag, alsook in het stroma, wat vervorming van de lamellaire architectuur veroorzaakt. De afzettingen geven een positieve immunohistochemische kleuring met antilichamen tegen keratoepitheline (2). De afzettingen verschijnen als amorfe roze afzettingen op hematoxyline en eosine (H&E) kleuring (zie figuur 1C en 1D) en kleuring met Congo rood kleuring waarbij de klassieke appelgroene birefringentie bij kruispolarisatie wordt aangetoond (zie figuur 2E en 2F) (1). Afwezigheid of dunner worden van de Bowman-laag, epitheliale atrofie en basale epitheliale degeneratie kunnen ook worden aangetroffen op histopathologisch onderzoek bij LCD (2).
LCD type I is de klassieke vorm van LCD die wordt veroorzaakt door een mutatie in het TGFβI-gen die resulteert in geïsoleerde amyloïde afzetting in de cornea. Er zijn vier LCD varianten geïdentificeerd: LCD type IIIA, type I/IIIA, type IV, en polymorfe amyloïdose. LCD-varianten presenteren zich later in het leven dan klassieke LCD. LCD type IIIA presenteert zich in het 5e-7e decennium, meestal met epitheliale erosies. Het heeft dikkere rasterlijnen, beschreven als “ropy-appearing”, die zich uitstrekken tot de limbus. LCD type I/IIIA heeft dunne rasterlijnen. LCD type IV presenteert zich in het 7e-9e decennium met kleine rasterlijnen. Amyloïde afzettingen in LCD type IV worden gevonden in het diepe stroma en epitheliale erosies komen zelden voor. Bij polymorfe amyloïdose type zijn de rasterlijnen afwezig en komen epitheliale erosies zelden voor (2).
LCD type II is een systemisch amyloïdose syndroom dat bekend staat als het Meretoja syndroom en de huid, de schedelzenuwen en het hoornvlies aantast. Het presenteert zich in de vroege volwassenheid met perifere neuropathieën, craniale neuropathieën, hound-achtig gezicht, droge huid, blepharochalasis, vooruitstekende lippen, en corneale tralielijnen. Dit type is in verband gebracht met het gelsolin-gen op chromosoom 9, dat codeert voor een amyloïde precursoreiwit dat actine verwijdert van plaatsen van verwonding en ontsteking (1). De naam is een verkeerde benaming en wordt niet beschouwd als een variant van lattice corneal dystrofie (2).
| A: Linkeroog bij retroilluminatie dat anterieure stromale afzettingen toont bij lattice corneal dystrofie | B: Linkeroog met hogere macht dat lineaire anterieure stromale afzettingen toont. |
 |
 |
| C: H&E vlek van cornea met rooster. Let op de roze amorfe afzettingen in het stroma | D: Een beter zicht op de roze, amorfe afzettingen |
 |
 |
| E: Kongo-rode kleuring, die amyloïd accentueert | F: Appelgroene birefringentie van amyloïd met kruispolarisatie. |
 |
 |
GRANULAIRE CORNEALE DYSTROPHY, TYPE I
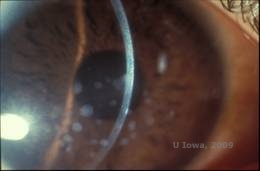
Granulaire corneale dystrofie, type I (GCD1) is een bilaterale, autosomaal dominante ziekte die geassocieerd is met een mutatie in het TGFβI-gen die leidt tot de afzetting van een hyalien materiaal in het hoornvliesstroma. De ziekte presenteert zich meestal vroeg in het eerste decennium van het leven met grijs-witte, “kruimel-achtige” troebelingen in het voorste tot middelste stroma, die zich uitbreiden naar het achterste stroma in gevorderde ziekte (1, 2). Deze opaciteiten zijn discrete afzettingen in het centrum, met helder hoornvlies in de periferie en helder hoornvlies tussen de afzettingen (zie figuur 3A en 3B). De ziekte is meestal asymptomatisch in een vroeg stadium, maar na verloop van tijd kunnen de troebelingen samengroeien en leiden tot verminderd gezichtsvermogen. Terugkerende cornea-erosies kunnen voorkomen bij GCD, maar met een lagere incidentie dan bij LCD (1, 5). Patiënten kunnen ook last hebben van verblinding en fotofobie (2). Behandeling in het begin van het ziekteproces is vaak alleen observatie. Naarmate de ziekte vordert, kunnen PTK en hoornvliestransplantatie echter nodig zijn om het gezichtsvermogen te verbeteren en de symptomen te doen verdwijnen. Net als LCD kan de ziekte terugkeren in hoornvliestransplantaties.
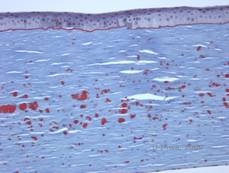
Histopathologisch zijn de troebelingen eosinofiele afzettingen die vaak worden beschreven als “rotsnoepachtig” in het anterieure stroma gemaakt van een hyalienachtig materiaal. Na verloop van tijd breiden de afzettingen zich uit naar het diepere stroma van het hoornvlies. Het hyalien materiaal kleurt helder rood met Masson trichroom kleuring (zie Figuur 3C en 3D).
| A: Spleetlampfoto van Granulaire Corneale Dystrofie, Type I | B: Let op de “kruimelachtige” stromale afzettingen met helder tussenliggend stroma. |
 |
 |
| C: H& E vlek van hoornvlies toont eosinofiele “kruimelachtige” hyaliene afzettingen in stroma | D: Hyalien materiaal kleurt helderrood met Masson-Trichrome |
 |
 |
GRANULAIRE CORNEALE DYSTROPHY, TYPE II
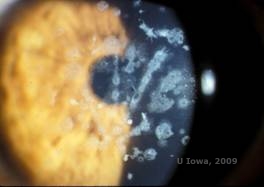
Granulaire corneale dystrofie, type II (GCD2), vroeger bekend als Avellino of gecombineerde korrel-rooster corneale dystrofie, is een autosomaal dominante ziekte die verband houdt met een mutatie in het TGFβI gen dat leidt tot een afzetting van zowel hyaline als amyloïd in het corneale stroma. Typisch presenteren patiënten zich in het tweede decennium van hun leven met kleine grijs-witte puntjes in het oppervlakkige stroma. De troebelingen kunnen er ook doornachtig, ringvormig of stervormig uitzien. Bij retroilluminatie zijn ze gedeeltelijk doorschijnend. Later in het ziekteproces kunnen zij ook tralielijnen ontwikkelen (zie figuur 4A en 4B). Deze lijnen kruisen elkaar niet en zien er witter en minder refractiel uit dan tralielijnen. Symptomen van GCD2 zijn pijn met epitheliale erosies en gezichtsstoornissen (2).
Histopathologisch zal de cornea stromale afzettingen vertonen die rood kleuren met Masson Trichrome, wat wijst op de aanwezigheid van hyaline (Zie figuur 4C). Bovendien zal de kleuring met Congo rood een appelgroene birefringentie vertonen bij kruispolarisatie, wat wijst op de aanwezigheid van amyloïd (zie figuur 4D). Men dacht dat de ziekte afkomstig was van een familie in Avellino, Italië. GCD type II is nu echter ook gemeld bij patiënten uit vele andere landen (2,5), met de hoogste prevalentie in Oost-Azië.
| A: Avellino dystrofie toont rasterachtige en korrelachtige afzettingen in cornea stroma | B. Masson Trichrome vlek die anterieure stromale hyaliene afzettingen toont |
 |
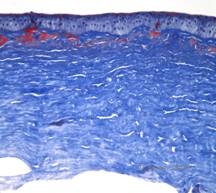 |
| C: Congo rood vlek die roze, amorfe amyloïde afzettingen toont in hetzelfde hoornvliesexemplaar. | D: Kruispolarisatie onthult appelgroene birefringentie die wijst op amyloïd. |
 |
 |
STROMALE CORNEALE DYSTROPHIEEN
MACULAIRE CORNEALE DYSTROPHIE
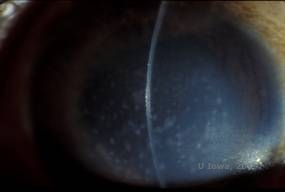
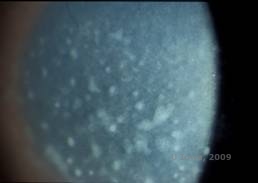
Maculaire corneale dystrofie (MCD) is een autosomaal recessieve ziekte die wordt veroorzaakt door een mutatie in het gen voor carbohydraatsulfotransferase 6 (CHST6) op chromosoom 16, die leidt tot een defect in de synthese van keratansulfaat, het belangrijkste glycosaminoglycan van het hoornvlies. De aandoening komt minder vaak voor dan LCD of GCD, maar heeft vaak ernstigere gevolgen voor het gezichtsvermogen. Hoewel MCD wereldwijd minder vaak voorkomt dan LCD of GCD, is het de meest voorkomende van de hoornvliesstromale dystrofieën op plaatsen zoals IJsland en Saoedi-Arabië (2,6). Grijs-witte, vlekachtige anterieure stromale laesies, vergelijkbaar met GCD1, verschijnen in het hoornvlies in het eerste decennium van het leven. In tegenstelling tot GCD1 is er echter stromale waas tussen de afzettingen en is vaak de hele cornea van limbus tot limbus betrokken (zie figuur 5A en 5B). Het hoornvlies is dun en naarmate de aandoening vordert wordt het Descemet membraan grijs en ontwikkelt guttae. Epitheliale erosies kunnen voorkomen, maar minder bij MCD dan bij LCD. Patiënten ontwikkelen meestal een ernstig verlies van gezichtsvermogen tegen het tweede of derde decennium van hun leven als gevolg van een diffuse troebeling van het hoornvlies. PTK kan worden uitgevoerd in sommige vroege gevallen van MCD. Deze aandoening is echter over het algemeen niet zo goed te behandelen met PTK als raster- of korreldystrofie en vereist vaak een hoornvliestransplantatie voor de behandeling (7). Recidief in transplantaten komt minder vaak voor bij MCD dan bij korrel- of korreldystrofie (1,2,5,6,8).
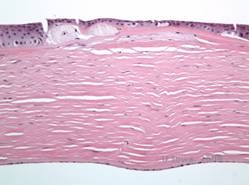
De stromale afzettingen bij MCD bestaan uit mucopolysacchariden die zich ophopen in het endoplasmatisch reticulum van keratocyten van het hoornvliesstroma, extracellulair tussen stromale lamellen, en binnen het epitheel, het Descemet membraan en het endotheel. Deze afzettingen kleuren blauw met Alcianblauw (zie figuur 5C en 5D) (1). Er zijn breuken in de Bowman laag en guttae met verdikking van het Descemet membraan (2).
Er zijn drie subtypes van MCD beschreven op basis van de aan- of afwezigheid van immunoreactief keratansulfaat in verschillende weefsels. Type I heeft geen immunoreactief keratansulfaat in het hoornvliesstroma, keratocyten, sera of kraakbeen, en is de meest voorkomende variant van MCD wereldwijd. Type IA heeft geen keratansulfaat in het stroma, sera en kraakbeen, maar wel aantoonbare niveaus in keratocyten. Type II heeft keratansulfaat in veel verlaagde gehalten in het stroma, de keratocyten, de sera en het kraakbeen (6).
| A: Spleetlampfoto van maculaire corneale dystrofie. | B: Let op de waas tussen de corneale stromale afzettingen |
 |
 |
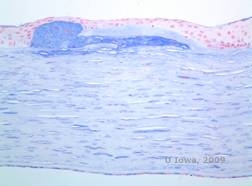
| C: H&E van cornea met maculaire dystrofie. Let op anterieure stromale afzettingen en verstoring van de Bowman’s laag | D: Mucopolysaccharide-afzettingen binnen keratocyten gemarkeerd met Alcian Blue-kleuring |
 |
 |
SCHNYDER CORNEAL DYSTROPHY (SCD)
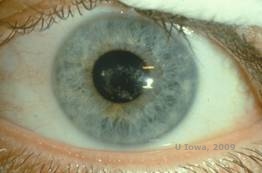
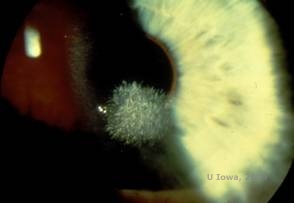
Schnyder corneal dystrofie (SCD), voorheen bekend als Schnyder kristallijne corneale dystrofie, is een autosomaal dominante, bilaterale corneale stromale dystrofie gekoppeld aan een genetische mutatie in UbiA prenyltransferase domain containing 1 (UBIAD1) gen op chromosoom 1. Het resulterende metabolische defect van corneale keratocyten leidt tot afzetting van kristallijne cholesterol in het stroma. De aanwezigheid van kristallen is echter niet absoluut noodzakelijk voor de diagnose van SCD. In feite heeft slechts 54% van de patiënten met SCD hoornvlieskristallen. Typisch presenteren patiënten zich in het tweede of derde decennium met een ringvormige centrale corneale troebelheid met of zonder kommavormige subepitheliale kristallen (zie figuur 6A en 6B). Vervolgens verschijnt arcus lipoides tussen de leeftijd van 23 en 38 jaar. Na de leeftijd van 38 jaar resulteert een progressieve vertroebeling van het hoornvlies in een panstromale waas tot in het midden van de periferie. De meeste patiënten ouder dan 50 jaar hebben fotopisch gezichtsverlies, verblinding en een verminderd gevoel van het hoornvlies, en daarom kan een chirurgische behandeling nodig zijn, inclusief hoornvliestransplantatie of PTK. Recidief in het transplantaat kan voorkomen. De ziekte is in verband gebracht met hypercholesterolemie, hyperlipidemie en genu valgum bij sommige patiënten (2,5,9,10).
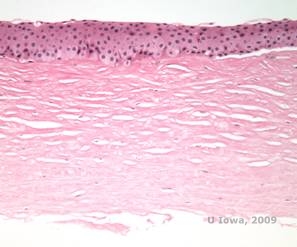
Histopathologisch zetten birefringente cholesterolkristallen, bestaande uit fosfolipiden en cholesterol, zich af binnen de basale epitheelcellen, keratocyten, de laag van Bowman, en tussen stromale lamellen. Lipiden lossen op bij normale histologische verwerking, dus moeten bevroren coupes van het hoornvlies worden verkregen om de aanwezigheid van lipiden aan te tonen met Oil-Red-O of Soedan zwartkleuring.
| A: Spleetlamp foto van Schnyder corneale dystrofie. | B. Centraal gelegen kristallijne afzettingen |
 |
 |
| C: H&E van cornea met SCCD | D. Oil Red O vlek benadrukt cholesterolkristallen die rood verschijnen. |
 |
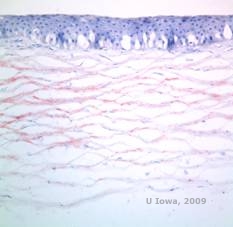 |
Tabel 4 biedt een gemeenschappelijk geheugensteuntje voor enkele van de hoornvliesdystrofieën die het stroma aantasten, de samenstelling van hun afzetting, en de methode om deze afzettingen te kleuren wordt opgesomd.
Tabel 4: Mnemonogram voor het onthouden van hoornvliesstromale dystrofieën
- Marilyn-Maculaire Dystrofie
- Monroe-Mucopolysaccharide
- Always-Alcian Blue vlek
- Gets-Granulaire Dystrofie
- Her-Hyaline
- Man in-Masson Trichrome vlek
- Los-Lattice Dystrofie
- Angeles-Amyloïd
- California-Congo Rood
OVERZICHT: CORNEALE STROMALE DYSTROPHIEEN
EPIDEMIOLOGIE
|
SIGNS
|
SYMPTOMEN
|
TREATMENT
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Conjunctivale en corneale pathologie. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3e editie. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D classification of corneal dystrophies–edition 2. Cornea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. Klinische en genetische aspecten van de TGFBI-geassocieerde corneale dystrofieën. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nature genet. 1994; 6: 47-51.
- Bron AJ. Genetica van de corneale dystrofieën: What we have learned in the past twenty five years. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating Keratoplasty for Macular Corneal Dystrophy. Ophthalmology 2005;112: 220-224.
- Badr IA, Wagoner MD. Phototherapeutic Keratectomy for Macular Corneal Dystrophy. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetics of anterior and stromal corneal dystrophies. Seminars in Ophthalmology, 2008; 23: 1,9-17.
- Weiss JS. Schnyder corneale dystrofie. Curr Opin Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. The gene for Schnyder’s crystalline corneal dystrophy maps to human chromosome 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Suggested citation format: Van C, Syed NA. Epithelial-Stromal and Stromal Corneal Dystrophies: A Clinicopathologic Review. Herziening van ; EyeRounds.org. August 20, 2015. Available from: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm