ABOVE: modified from © ISTOCK.com, tera vector
Etwas aufzubauen ist fast immer schwieriger als es abzureißen. Genauso ist es eine größere Herausforderung, Gene einzuschleusen als sie auszuschalten. Das ist eine Realität, die Forscher überwinden müssen, um das Beste aus der Genbearbeitung herauszuholen. Das Knocking-in von Genen ermöglicht es den Wissenschaftlern, die Auswirkungen spezifischer Genvarianten zu untersuchen, Reportergene wie das grün fluoreszierende Protein zu verwenden, um Genprodukte zeitlich und räumlich zu verfolgen, die Genomregulierung zu untersuchen und schließlich krankheitsverursachende Gene zu reparieren. „Es ist ein wirklich effektiver Weg, jede Base eines Gens zu untersuchen“, sagt Greg Findlay, Doktorand an der University of Washington.
CRISPR-Cas9, eine für ihre Benutzerfreundlichkeit bekannte Gene Editing-Technologie, kann Gene ein- oder ausschalten. Um ein Gen auszuschalten, wird CRISPR-Cas9 mit Hilfe einer Leit-RNA, die das Werkzeug auf das gewünschte Gen ausrichtet, in eine Zelle eingebracht. Dort schneidet Cas9 das Gen, indem es beide DNA-Stränge durchtrennt, und der reguläre DNA-Reparaturmechanismus der Zelle repariert den Schnitt durch einen Prozess, der als nicht-homologes Endjoining (NHEJ) bezeichnet wird. NHEJ ist hocheffizient, aber ungenau. Der Prozess neigt dazu, Fehler in Form von kleinen Insertionen oder Deletionen einzuführen, die normalerweise ausreichen, um das Gen auszuschalten.
Um ein Gen einzuschalten, müssen die Schnitte jedoch sehr präzise repariert werden, ohne zusätzliche Insertionen oder Deletionen. Dies erfordert einen zweiten DNA-Reparaturmechanismus, die so genannte homologiegeleitete Reparatur (HDR), die – zumindest in Säugetierzellen – weniger effizient abläuft, so dass ihre Häufigkeit hinter der von NHEJ zurückbleibt. Erschwerend kommt hinzu, dass einige Genorte und Zelltypen von Natur aus weniger geeignet für die CRISPR-Cas9-Bearbeitung sind.
In den letzten Jahren haben Forscher viele neue Strategien entwickelt, um die Effizienz der CRISPR-Cas9-Bearbeitung von großen und kleinen Genen zu steigern, und sie haben neue Anwendungen für diese Art der Genbearbeitung vorgeschlagen und getestet. The Scientist stellt hier einige der vielversprechendsten Ansätze vor.
Wählen Sie es aus
Forscher: Jon Chesnut, Senior Director of Synthetic Biology R&D, Thermo Fisher Scientific
Projekt: Bei der Entwicklung eines Gen-Tagging-Kits namens Truetag, das Thermo Fisher später in diesem Jahr auf den Markt bringen wird, verwendete Chesnut selektierbare Marker, um die
Effizienz zu verbessern. Ein selektierbarer Marker – in diesem Fall ein Antibiotikaresistenzgen – wird mit einem fluoreszierenden Protein verbunden und in Säugetierzellen eingeschleust. Diese Zellen werden dann in Kulturen mit dem entsprechenden Antibiotikum gezüchtet. Das Resistenzgen verschafft den Zellen, die es tragen, einen Selektionsvorteil; nur sie sind in der Lage zu wachsen, und die Zellen, die wachsen, enthalten die gewünschte Genmarkierung. Selbst wenn die Effizienz der Geneinfügung gering ist, können die Forscher die Antibiotikaselektion für eine Woche oder länger anwenden, um einen hohen Prozentsatz von Zellen mit erfolgreicher Einfügung zu erhalten.
Bei Verwendung des Antibiotikums Puromycin oder Blasticidin mit dem Kit gelang es Chesnuts Team, die Geneinfügungsrate von 10-30 Prozent auf 90 Prozent oder mehr in einigen Zellpopulationen zu erhöhen. Bei einigen besonders schwierigen Genen stieg die Einbaurate von weniger als 1 Prozent auf über 90 Prozent. Es ist wichtig, mehrere Dosen von Antibiotika an der Zelllinie zu testen, die man verwenden will, um die richtige Dosis zu finden, sagt Chesnut: Man will Zellen ohne Einfügungen abtöten, aber nicht Zellen mit erfolgreichen Einfügungen.
Try It: Selektierbare Marker funktionieren am besten, wenn das betreffende Gen stark exprimiert wird, sagt Chesnut. „Ist das nicht der Fall, kann man zwar eine Selektion erreichen, aber die Expression des fluoreszierenden Proteins ist nicht ausreichend, um es zu erkennen.“ Außerdem gelten die allgemeinen Einschränkungen von CRISPR-Cas9. „Es gibt Regionen des Genoms, die sich mit CRISPR nicht so gut schneiden lassen, und wir wissen immer noch nicht genau, warum“, fügt er hinzu. Und einige Zelltypen nehmen fremde DNA, RNA oder RNA-Protein-Komplexe – die drei Methoden der CRISPR-Cas9-Übertragung – nicht ohne Weiteres an.
Wenn Sie selektierbare Marker einfügen möchten, sollten Sie darauf achten, dass sich eine so genannte PAM-Sequenz, eine kurze Markierung in der Ziel-DNA, die CRISPR-Cas9 erkennen muss, bevor es schneidet, innerhalb von 10 Basenpaaren der gewünschten Geneinfügungsstelle befindet, sagt Chesnut. Liegt die PAM-Sequenz weiter von der Schnittstelle entfernt, ist die Einfügeeffizienz möglicherweise zu gering, um funktional zu sein. Ohne eine PAM-Stelle kann man es mit TALENs oder Zinkfingernukleasen versuchen, obwohl diese älteren Geneditierungstechniken schwieriger sind als CRISPR.
Timed Inhibition
Forscher: Jacob Corn, Genombiologe, Eidgenössische Technische Hochschule, Zürich
Projekt: Forscher verstehen nicht, warum der NHEJ-Stoffwechselweg den HDR-Stoffwechselweg in Säugetierzellen bei weitem übertrumpft. „Hefezellen machen HDR wie verrückt“, sagt Corn. In dem Bestreben, diesen DNA-Reparaturprozess in menschlichen Zellen zu beschleunigen und die Kontrolle über die Knock-in-Gene zu verbessern, versuchen er und sein Team herauszufinden, wie HDR reguliert wird. Sie untersuchten menschliche Zellen auf Gene, deren Knockdown zu einer erhöhten HDR in der Zelle führte, und suchten dann nach kleinen Molekül-Inhibitoren für diese Gene. Eines der gefundenen Gene kodiert für CDC7, eine Kinase, die den Übergang in die S-Phase des Zellzyklus reguliert. Sein Inhibitor, XL413, steigerte die Effizienz des Gen-Knock-In um das Zwei- bis Dreifache (BioRXiv, DOI: 10.1101/500462, 2018). Das liegt daran, dass HDR nur in einigen Teilen des Zellzyklus stattfindet, einschließlich der S-Phase, sagt Corn. Wenn man den Inhibitor XL413 zur gleichen Zeit hinzufügt, in der man CRISPR-Cas9 verwendet, um sein Zielgen zu bearbeiten, stapeln sich die Zellen in der Phase unmittelbar vor der S-Phase. Entfernt man dann XL413, gehen alle Zellen in die S-Phase und erhöhen die Knock-in-Effizienz.

Corn hat diese Technik bei vielen unsterblichen menschlichen Zelllinien und bei menschlichen T-Zellen eingesetzt. Sie kann sowohl kurze Abschnitte der DNA, wie SNPs, als auch große Gene verändern. Es gibt keinen Grund, warum es bei Mäusen nicht funktionieren sollte, sagt er, obwohl er es nicht getestet hat.
Versuchen Sie es: „Das Timing ist absolut entscheidend“, sagt Corn. Cas9 muss die DNA zur gleichen Zeit schneiden, zu der XL413 hinzugefügt wird. Wenn Sie beim Editieren mit CRISPR-Cas9 zuerst hemmen und dann freisetzen, sinkt die Effizienz der homologen Rekombination um das Dreifache, anstatt zu steigen, weil die Zellen in der falschen Phase des Zellzyklus freigesetzt werden.
Und wie bei jeder HDR-Maßnahme, sagt Corn, führen Sie immer eine Kontrolle ohne Nuklease durch, um sicherzustellen, dass Sie nicht versehentlich kontaminierte DNA amplifizieren, die in Ihrem Labor herumschwirrt. Nach der Einführung des Knock-in gilt: Sequenz, Sequenz, Sequenz, Sequenz“, sagt er. Die Verwendung eines Reportersystems, wie z. B. eines fluoreszierenden Protein-Tags, zum Nachweis der erfolgreichen Geneinfügung kann nach hinten losgehen. Die Sequenzierung verifiziert, dass die Einfügungen an der richtigen Stelle vorgenommen wurden.
Playing the Long Game
Forscher: Channabasavaiah Gurumurthy, Direktor der Maus-Genom-Engineering-Kerneinrichtung, University of Nebraska Medical Center
Projekt: Als Gurumurthy und seine Kollegen vor einigen Jahren über die Schwierigkeit nachdachten, Gene in Maus-Zygoten einzuschleusen, hatten sie eine Offenbarung.
Die Forscher fügten erfolgreich kurze, einsträngige DNA ein, warum also nicht versuchen, ein Knock-in durch Einfügen von langer, einsträngiger DNA zu erreichen? Tatsächlich steigert der Ansatz, den Gurumurthy Easi-CRISPR (efficient additions with ssDNA inserts -CRISPR) nennt, die Effizienz um das 2,5-fache, und die Verwendung von einzelsträngiger DNA senkt die Rate der Off-Target-Insertionen in Zellkulturen um das 100-fache (Nat Protoc 13:195-215, 2018; Nature 559:405-09, 2018). „Das ist ziemlich gewaltig“, sagt er. In Gurumurthys Labor hat Easi-CRISPR eine Knock-in-Mauslinie für 9 von 10 Genen erzeugt, die sie getestet haben. Ein Mitarbeiter hat es auch bei menschlichen T-Zellen eingesetzt, um CAR-T-Zellen zu erzeugen, patientenspezifische Immunzellen zur Krebsbekämpfung.
Versuchen Sie es: Easi-CRISPR ist bei weitem nicht narrensicher, warnt Gurumurthy. Manchmal fügt die Technik nur einen Teil des Gens ein. Außerdem, so fügt er hinzu, können die Homologiearme – die kurzen Sequenzen auf beiden Seiten des Gens, die es zu seinem richtigen Ziel im Genom führen – durcheinander geraten. Und einige Loci sind unerklärlicherweise schwieriger einzufügen als andere.
Nur wenige kommerzielle Anbieter entwerfen und synthetisieren maßgeschneiderte lange, einsträngige DNA. Sie können Ihre eigene herstellen, aber die Stabilität der einzelsträngigen DNA variiert; weniger stabile Sequenzen haben eine geringere Ausbeute, so dass Sie möglicherweise mehr von ihnen synthetisieren müssen, sagt Gurumurthy.
Forscher, die nicht in der Lage sind, CRISPR in einzellige Mausembryonen einzufügen, können eine Kerneinrichtung dafür bezahlen, die Mäuse mit ihrer DNA-Sequenz herzustellen, sagt Gurumurthy. Kerneinrichtungen wie die seine verlangen zwischen 5.000 und 15.000 Dollar für die Erzeugung von ein oder zwei Zuchtpaaren; kommerzielle Einrichtungen verlangen 20.000 bis 50.000 Dollar, sagt er.
Knock-in nach Zahlen
Forscher: Greg Findlay, MD/PhD-Kandidat im Labor von Jay Shendure, University of Washington
Projekt: Findlay und seine Kollegen wollten die Interpretation von Mutationen im Brust- und Eierstockkrebsgen BRCA1 durch Kliniker verbessern. Dieses Gen weist Tausende von Varianten auf, aber die Forscher wissen nicht, wie die meisten von ihnen seine Funktion beeinflussen. Um die Auswirkungen dieser Varianten zu untersuchen, verwendeten sie eine von ihnen entwickelte Knock-in-Technik, die als Sättigungs-Genom-Editierung bezeichnet wird (Nature, 562:217-22, 2018).
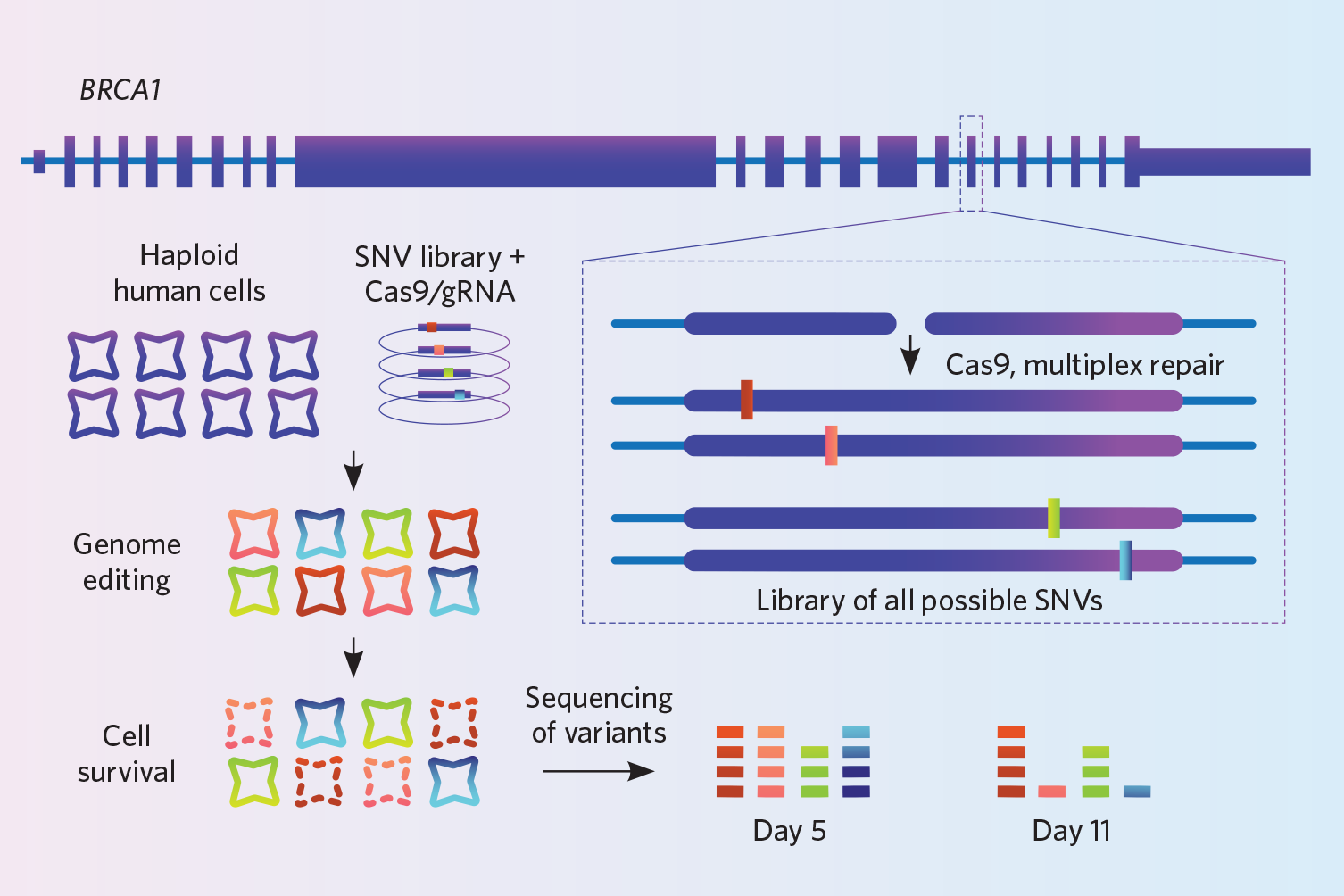
In einer immortalisierten haploiden menschlichen Zelllinie verwendeten sie CRISPR-Cas9, um 4.000 winzige Varianten in Millionen von Zellen gleichzeitig in vitro einzuschleusen. Das Genom wird in jeder Zelle an der gleichen Stelle geschnitten, aber das Genom jeder Zelle erhält eine andere Variante. Um die HDR zu fördern, schalteten sie auch das Ligase4-Gen aus und deaktivierten damit den NHEJ-Reparaturweg – ein Schritt, der laut Findlay zu einer dreifachen Effizienzsteigerung führte. Da alle Knock-Ins der Zellen unterschiedlich sind, sequenzierten sie die Zellen gründlich, indem sie dieselbe genomische Region millionenfach abdeckten, um sicherzugehen, dass sie tatsächlich die 4.000 Varianten, die sie untersuchen wollten, eingeknockt hatten. Sie sequenzierten zu zwei Zeitpunkten und folgerten daraus, dass die Knock-ins, die bei der Sequenzierung zum zweiten Zeitpunkt nicht auftauchten, diejenigen waren, die die Funktion des Gens beeinträchtigten, weil die Zellen, die sie trugen, gestorben sein mussten.
Versuchen Sie es: Findlays Team ließ sich die DNA-Oligos für die 4.000 Varianten auf einem Microarray herstellen. Man kann Arrays mit 6.000 bis 250.000 Oligos kaufen, also sollte man in Erwägung ziehen, mehr für sein Geld zu bekommen, indem man mehrere Experimente auf demselben Array kombiniert, sagt Findlay. Ihr Labor zahlt etwa 5.000 Dollar für 100.000 Oligos.
Diese Strategie hat ihre Grenzen: Sie wurde bisher nur für das Klopfen von Einzelnukleotidvarianten verwendet, und alle Änderungen müssen im selben Gen vorgenommen werden. Die Methode funktioniert am besten, wenn eine relativ schmale DNA-Region bearbeitet wird, etwa 110-120 Basenpaare, da längere DNA-Oligos zu viele Fehler aufweisen würden, so Findlay. Es ist auch wichtig, sehr tief zu sequenzieren, um sicherzustellen, dass die gesamte Anzahl der Varianten, die man einfügen möchte, berücksichtigt wird.