ABOVE: modified from © ISTOCK.com, tera vector
ほとんど常に、何かを作ることはそれを壊すことよりも難しいのです。 同様に、遺伝子をノックインすることは、ノックアウトすることよりも大きな挑戦となります。 遺伝子編集を最大限に活用するためには、研究者が乗り越えなければならない現実なのです。 遺伝子をノックインすることで、特定の遺伝子変異の影響を調べたり、緑色蛍光タンパク質などのレポーター遺伝子を用いて遺伝子産物を時間的・空間的に追跡したり、ゲノムの制御を調べたり、最終的には病気の原因となる遺伝子を修復したりすることができる。 ワシントン大学の医学博士候補であるグレッグ・フィンドレー氏は、「これは、遺伝子のすべての塩基を調べるのに実に効果的な方法です」と述べています。
CRISPR-Cas9 は、その使いやすさで知られる遺伝子編集技術で、遺伝子をノックインまたはノックアウトできます。 遺伝子をノックアウトするには、目的の遺伝子にツールを向けるガイドRNAを使ってCRISPR-Cas9を細胞内に挿入する必要があります。 そこでCas9が遺伝子を切断し、DNAの両鎖を切り取る。細胞の通常のDNA修復機構は、非相同末端結合(NHEJ)と呼ばれるプロセスを用いて切断箇所を修復する。 NHEJは非常に効率的であるが、不正確である。 しかし、遺伝子をノックアウトするためには、切断部分を非常に正確に修復し、余分な挿入や欠失がないようにしなければならない。 このためには、相同性指向性修復(HDR)と呼ばれる第2のDNA修復機構を利用する必要があるが、少なくとも哺乳類細胞では、この機構はあまり効率よく発生しないため、その頻度はNHEJのそれに比べて小さい。 このプロセスをさらに複雑にしているのは、一部の遺伝子座や細胞型が、もともとCRISPR-Cas9編集に適していないという事実です。
過去数年間、研究者は、CRISPR-Cas9を使って大小さまざまな遺伝子をノックインする効率を高めるための多くの新しい戦略を開発し、その過程で、このタイプの遺伝子編集の新しいアプリケーションを提案し検証してきました。 ここでは、The Scientistが最も有望なアプローチのいくつかを探ります。
Select It
研究者。 Jon Chesnut, senior director of synthetic biology R&D, Thermo Fisher Scientific
Project: サーモフィッシャー社が今年後半に発売する「Truetag」という遺伝子タギングキットの開発において、チェスナット氏は選択可能なマーカーを用いて
効率を向上させた。 選択可能なマーカー(この場合は抗生物質耐性遺伝子)を蛍光タンパク質のタグに貼り付け、
哺乳類の細胞にノックします。 その細胞は、関連する抗生物質とともに培養される。 耐性遺伝子は、それを持つ細胞に選択的優位性を与え、その細胞だけが成長することができ、したがって成長した細胞は目的の遺伝子タグを含む。 遺伝子挿入の効率が低くても、研究者は1週間以上抗生物質を選択することで、挿入に成功した細胞の割合を高くすることができる。
このキットで抗生物質ピューロマイシンまたはブラストサイジンを使用すると、Chesnut氏のチームは、いくつかの細胞集団で遺伝子挿入率を10~30%から90%以上まで高めることに成功した。 特に難しい遺伝子は、挿入率が1パーセント以下だったのが90パーセント以上になった。 挿入のない細胞は殺したいが、挿入に成功した細胞は殺したくないのです」
Try It.のように、使用予定の細胞株で複数回の抗生物質の投与を試し、正しい投与量を見つけることが重要である、とChesnutは言う。 選択マーカーは、目的の遺伝子が高度に発現しているときに最も効果的であると、Chesnut氏は言う。 「そうでない場合、選択マーカーはまだ使えるかもしれないが、蛍光タンパク質タグを検出するのに十分な発現が得られないかもしれない」。 また、CRISPR-Cas9の一般的な限界も適用されます。 “ゲノムには、CRISPRであまりうまく切れない領域があり、その理由はまだ分かっていません。”と彼は付け加えます。 そして、細胞の種類によっては、外来DNA、RNA、RNA-タンパク質複合体(CRISPR-Cas9の3つの送達方法)を容易に受け入れないものもあります。
選択マーカーをうまく挿入するには、いわゆるPAM配列(CRISPR-Cas9が切断前に認識すべき標的DNAの短いタグ)が、目的の遺伝子挿入部位から10塩基対以内にあることを確認すると、Chesnut博士は述べています。 これ以上切断部位から遠ざかると、挿入効率が低すぎて機能しなくなる可能性があります。 PAMサイトがなければ、TALENやジンクフィンガーヌクレアーゼを試すことができますが、それらの古い遺伝子編集技術はCRISPRよりも厄介です。
Timed Inhibition
研究者。 Jacob Corn, genome biologist, Swiss Federal Institute of Technology, Zurich
Project: 哺乳類細胞において、なぜNHEJ経路がHDR経路に圧倒的に勝るのか、研究者は理解していない。 「酵母は狂ったようにHDRを行います」とCorn氏は言う。 ヒト細胞でこのDNA修復プロセスを活性化し、遺伝子ノックイン制御を改善するために、コーン博士と彼のチームは、HDRがどのように制御されているかを突き止めようと試みている。 彼らは、ヒト細胞をスクリーニングして、ノックダウンによって細胞内のHDRが増加する遺伝子を探し出し、その遺伝子の低分子阻害剤を探した。 飛び出した遺伝子の1つは、細胞周期のS期への移行を制御するキナーゼであるCDC7をコードしており、その阻害剤であるXL413は、遺伝子ノックイン効率を2~3倍に高めた(BioRXiv, DOI: 10.1101/500462, 2018)。 それは、HDRがS期を含む細胞周期の一部でしか起こらないからだと、Cornは言う。 CRISPR-Cas9を使って標的遺伝子を編集するのと同時に阻害剤XL413を加えると、S期直前の段階で細胞が積み上がるのです。

また、あらゆるHDR作業と同様に、ラボに漂っている汚染DNAを誤って増幅しないように、常に無ヌクレアーゼ制御を実行すると、Corn氏は述べています。 ノックインを導入した後は、「シーケンス、シーケンス、シーケンス、シーケンス」と言う。 蛍光タンパク質タグのようなレポーターシステムを使って、遺伝子挿入が成功したことを証明するだけでは、逆効果になりかねません。 配列決定により、正しい部位に挿入されたことが確認される。 Channabasavaiah Gurumurthy、ネブラスカ大学医療センター、マウスゲノム工学コア施設ディレクター
Project: 数年前、マウス接合体に遺伝子をノックインすることの難しさについて考えていたところ、Gurumurthy氏と彼の同僚は、ある啓示を受けました。
研究者たちは短い一本鎖のDNAを挿入することに成功していたのですから、長い一本鎖のDNAを挿入してノックインを試みてはどうでしょうか。 実際、GurumurthyがEasi-CRISPR(efficient additions with ssDNA inserts -CRISPR)と呼ぶこのアプローチは、効率を2.5倍に高め、一本鎖DNAを使うことで細胞培養における標的外挿入率を100倍に削減します(Nat Protoc 13:195-215, 2018; Nature 559:405-09, 2018)。 “かなり巨大です “と彼は言います。 Gurumurthyの研究室では、Easi-CRISPRは、彼らが試した10個の遺伝子のうち9個についてノックインマウス系統を生成しています。 共同研究者はまた、癌と戦うための患者特異的な免疫細胞であるCAR-T細胞を作るために、ヒトのT細胞でこれを使用しました。 Gurumurthy氏は、Easi-CRISPRは完全無欠とは言い難いと警告している。 この技術では、遺伝子の一部しか挿入されないこともある。 また、相同性アーム(遺伝子の両側にある短い配列で、ゲノムの正しいターゲットに遺伝子を導くもの)を壊してしまうこともある、と彼は付け加えている。 そして、ある遺伝子座は、他の遺伝子座よりも挿入するのが不可解なほど難しいのです」
長い一本鎖のカスタムDNAを設計、合成している商用ベンダーはほとんどありません。 自分で作ることもできますが、一本鎖DNAの安定性はさまざまです。安定性の低い配列は収量が少なく、より多くの本数を合成する必要があるかもしれないと、Gurumurthy氏は言います。 彼のような中核施設は、1~2組の繁殖ペアを作るのに5000~15000ドル、商業施設は2~5万ドルかかると彼は言います。
Knock-in By Numbers
Researcher: Greg Findlay, MD/PhD candidate in the lab of Jay Shendure, University of Washington
Project: フィンドレー氏と彼の同僚は、乳がんおよび卵巣がん遺伝子BRCA1における変異を臨床医が解釈する方法を改善することを目的としていました。 この遺伝子には何千もの変異があるが、研究者たちはそのほとんどがその機能にどのような影響を及ぼすのか分かっていない。 これらの変異体の影響を研究するために、彼らは飽和ゲノム編集と呼ばれる彼らが開発したノックイン技術を使用しました(Nature, 562:217-22, 2018)
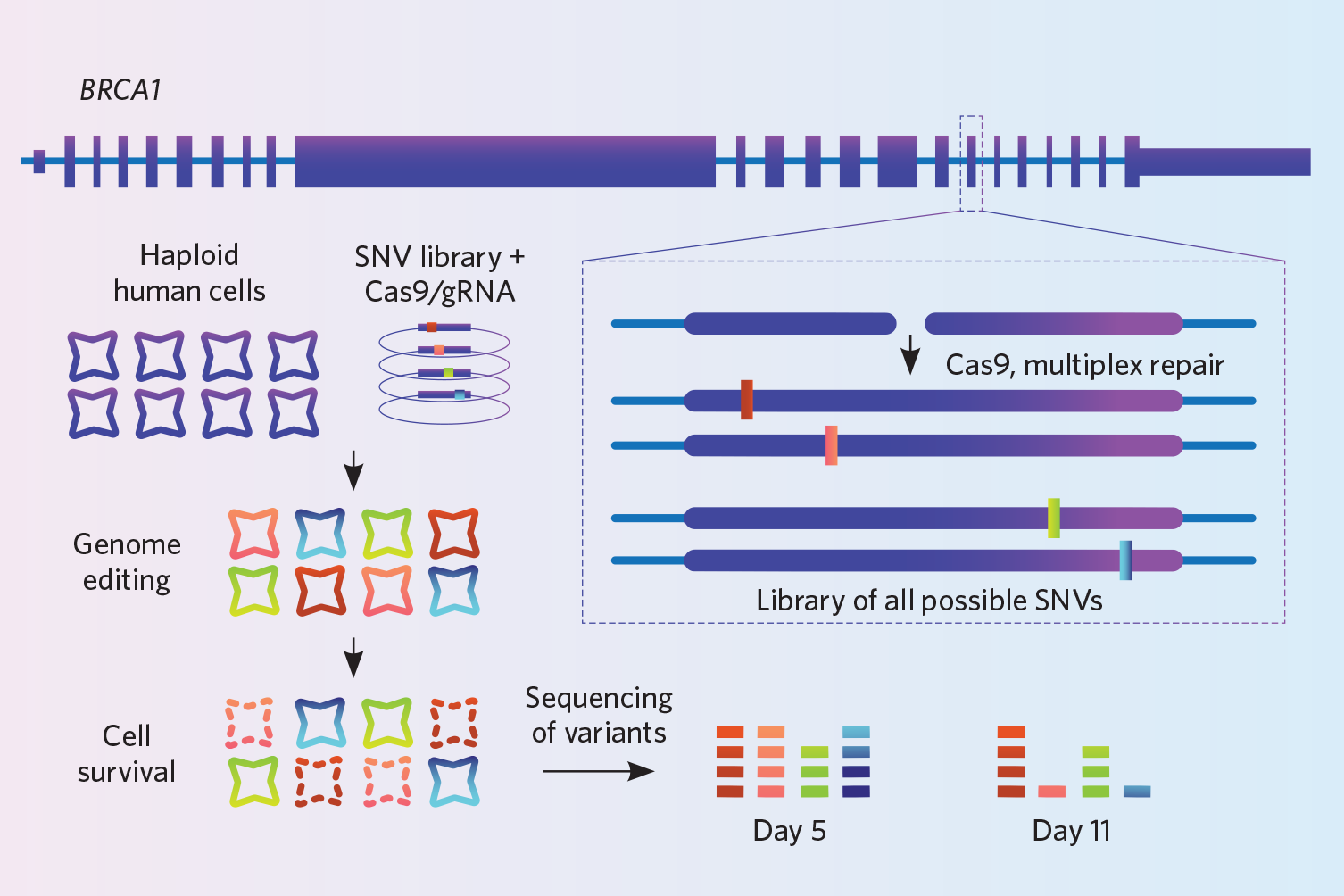
Try It: フィンドレーのチームは、4,000のバリアントのDNAオリゴをマイクロアレイで製造してもらいました。 6,000から250,000個のオリゴを含むアレイを購入することができますので、同じアレイで複数の実験を組み合わせて、より多くの利益を得ることを検討してください、とフィンドレーは言います。 この戦略には限界がある。今のところ、1塩基の変異体をノックインするためにしか使われていないし、編集はすべて同じ遺伝子で行わなければならない。 この方法は、110~120塩基対程度のかなり狭い範囲のDNAを編集する場合に最適です。なぜなら、これより長いDNAオリゴではエラーが多すぎるからです、とフィンドレー氏は言います。 また、ノックインしようとする変異体の数を完全に網羅するために、非常に深く配列することも重要である。