背景
カリウムは周期表のアルカリ金属族に属する銀白色の柔らかい高反応性陽イオンであり、その性質から、高カリウム血症、ナトリウム・カリウムポンプ、心臓の働きが重要視されています。
欧米人の食事には平均して1日あたり80~100mEqのカリウムが含まれており、通常の生理的条件下では、その90%が受動的に吸収され、わずか9.0mmolが糞便中に排泄されるにとどまる。 体内に蓄積された3500-4000mmolは、夜間と早朝に最も低く、午後の時間帯に最も高くなるという厳しいホメオスタシス機構によって通常3.5-5.3mmol/Lの範囲に維持されている日中の血漿カリウムレベルには不釣り合いである。
一旦血流に吸収されると、カリウムの摂取量と排出量を一致させるのが腎臓の役割となり、数時間かかります。その間、インスリンとカテコールアミンの影響下で「内部カリウムバランス」が、カリウムを細胞内と細胞外の空間の間で移動させることにより、一時的に恒常性を維持するのです。 α受容体を刺激するとカリウムの細胞内への侵入が阻害され、β受容体を刺激するとナトリウム・カリウムATPアーゼポンプの活性化によりカリウムの侵入が促進される。
ナトリウム・カリウムATPアーゼポンプはサルコレマにあるゲートキーパー酵素である。 細胞内に保持されているカリウムの98%(約144.0mmol)を保護するのに役立っています。 これにより、特に神経細胞や心筋細胞などの興奮性の細胞で、適切な細胞機能に必要な細胞膜の重要な電位差を維持することができるのです。
カリウムの正常な生理と病態生理
カリウムは急速に吸収された後、インスリンとアルドステロンの放出を通じて、自身の体内レベルを調整するのを助けます。 その他、β2アドレナリン受容体、アルカリ性血液のPH、細胞の同化作用など、カリウムの体内濃度を制御する固有の身体刺激も見出されています。
インスリンとアルドステロンの放出。 摂取されたカリウムは速やかに循環に入る。 門脈循環に到達すると、膵臓を刺激してインスリンを分泌させる。 同時に、循環中のカリウムは次糸球体細胞に到達し、レニンが放出される。 レニンは肝臓に到達するとアンジオテンシンIに変換され、アンジオテンシンIは肺に移動し、アンジオテンシンIIに変換される。 アンジオテンシンIIは、循環血液を介して腎臓に戻り、糸球体座を刺激してアルドステロンを分泌させる<5352>。 食後に分泌されたインスリンは主に骨格筋に作用し、グルコーストランスポーターGLUT4の挿入を担うAKT依存性経路と、細胞内のナトリウムカリウムATPaseを活性化してカリウムを細胞内空間に移行させるAPK経路の2つを活性化させる。 AKT依存性経路とは異なり、APK経路はメタボリックシンドロームや慢性腎臓病でも障害されない(図1)。 腎糸球体でろ過されたカリウムは、供給されたナトリウムと水の量に比例して、近位尿細管とヘンレのループで受動的に再吸収される。 通常、濾過された負荷量の約10%しか遠位ネフロンに到達しない。
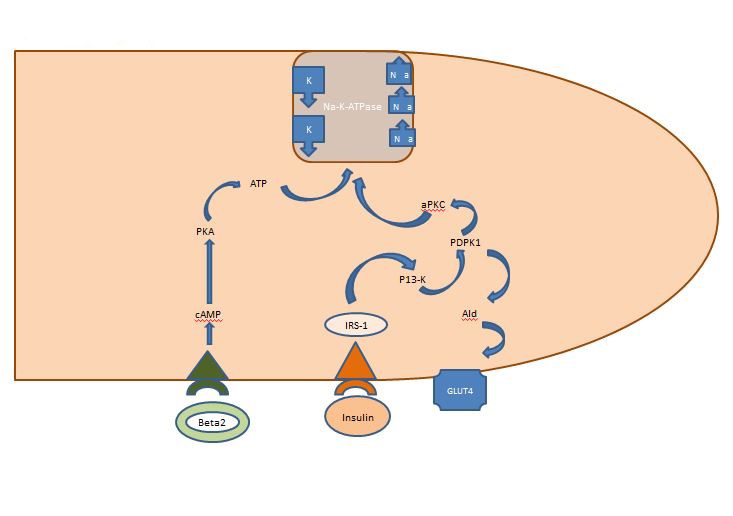
図1. 骨格筋細胞に対するインスリンの作用。 食後に放出されたインスリンは、骨格筋において、グルコーストランスポーターGLUT4の挿入を担うAKT依存経路と、細胞内のナトリウムカリウムATPaseを活性化してカリウムを細胞内空間に移行させるAPK経路の2つの経路を活性化する。
遠位輸液管の始まりで過剰カリウムの分泌が始まり、さらに遠位ネフロン、集合管に進むにつれ徐々に増加する。 これは、α-インターカレーション細胞上の水素カリウムATPaseのアップレギュレーションによって媒介される。
腎臓の管周囲細胞でカリウム濃度が高くなると、RAAS系が活性化してアルドステロンを放出し、これが基底膜のナトリウムカリウムATPaseを活性化して細胞内のナトリウムを減少させ、膜電圧を過分極させてカリウムの取り込みを電気泳動的に増加させて尿中に排泄できるようになる .
高カリウム血症では、結腸から排泄されるカリウムの量が最大で30%増加することがあります。 腎不全の場合、カリウムは大腸腸管細胞の基底膜にある活性化ナトリウムカリウムATPaseポンプによって活発に取り込まれ、細胞の頂膜のカルシウム依存性大カリウムチャネルを通って、反対側の大腸内腔に排泄されます。
以上のことから、カリウムの血漿レベルの恒常性のメカニズムは、主に3つの同時取引-カリウムの摂取、カリウムの細胞内/細胞外シフト、カリウムの尿中排泄-の相互作用によって規定されており、これらはすべて最終的にはナトリウムカリウムポンプに依存していることが判る。
高カリウム血症による差し迫った危険のメカニズムとその管理を理解するためには、活動電位の電気生理学とナトリウム・カリウムATPアーゼ酵素の内部を理解する必要があります。 イオンが濃度勾配を下る「化学電位」とイオンや分子が電荷を反発させる「電気電位」の2つの電位の差によって決定され、膜貫通電位(TMP)が生じ、+側イオンが細胞の外側へ、またはその逆の純移動をするとき+側と言われる。
非ペースメーカー心筋細胞の活動電位
活動電位には5つの相があり、第4相で始まり、第4相で終わる。 このプロセスに関与するポンプは、サルコレマ・ナトリウム・カルシウム交換体、カルシウムATPアーゼ、そして最終的にはナトリウム・カリウムATPアーゼである。
- フェーズ4。 静止期:これは内向き整流子チャネルを介してカリウムが常に外向きに移動する結果、静止電位が-90mVとなるものである。 第0相:ペースメーカー細胞の発火や隣接する細胞との伝導により、TMPが-90mV以上に上昇することで脱分極が始まる。 この時点で「高速ナトリウムチャネル」が次々と開き始め、細胞内にナトリウムが入り込んでTMPが上昇し、十分な高速ナトリウムチャネルが開いて-70mVになると、自立的な内向きナトリウム電流が動き出す。 TMPを0mVまで急速に脱分極させ、「オーバーシュート」として知られる一時的な間を過ごす。この時点で、時間依存の高速ナトリウムチャネルが閉じ、「長開放」カルシウムチャネルが開いてTMPを-40mVまで引き上げ、その濃度勾配を下る小さな定常カルシウム流入を可能にする。
- フェーズ1. 初期再分極期:TMPがわずかに上昇し、いくつかのカリウムチャネルが短時間開いた結果、カリウムが細胞の外側に流れ、TMPが約0mVに戻ることから始まります。 プラトー期:ここでは2つの逆電流が電気的にバランスし、その結果、TMPは0mVのすぐ下でバランスした状態に維持される。 “長く開いているカルシウムチャネル “はまだ開いており、その結果、細胞内に一定のカルシウムが流れ込む。 遅延整流カリウムチャネルは、その濃度勾配を下ってカリウムを細胞の外側に通過させる。
- 第3相。 再分極期:この段階では、カルシウムチャネルは徐々に不活性化され、細胞外へのカリウムの持続的な流れは、このように内向きのカルシウムの流れを上回り、カリウムを細胞内空間に、ナトリウムとカルシウムを細胞外に戻す。
心臓ペースメーカー細胞の活動電位
心臓ペースメーカー細胞には生来の自動性があるため、リズミックサイクルで脱分極することができる。 心房結節(SAN)が60~90/minと最も高い自己分極リズムを持ち、次いで房室結節(AVN)が40~60/min、そしてプルキンエ線維と心室筋が20~40/minである。
ペースメーカー細胞の膜電位は不安定で、活動電位の位相は明確でない。 ペースメーカー細胞は内向き整流カリウムチャネルが少なく、そのTMPは-60mV以下に低下しないため、-90mVのTMPを必要とする高速ナトリウムチャネルの役割はなく、結果として急速脱分極相が存在しないことになった。
TMPが>-60mVになると、遅いナトリウムチャネルを通してイオンが自然に流れ、TMPが<-50mVまで脱分極し、カルシウムチャネルが閉じると再び-60mVに戻る「おかしな/ペースメーカー」電流が作動する。
不応期
第2相の長いプラトー中の遅いカルシウムチャネルによる長い不応期は、次の収縮の前に心室を完全に空にするために必要な時間を提供する。 不応期には、絶対的不応期(ARP)、有効不応期(ERP)、相対的不応期(RRP)がある。 ARPでは、細胞は絶対に興奮しない。
ERPはARPから第3相の短いセグメントまで続く。 この時点での刺激は細胞を最小限度に脱分極させるが、そのレベルは近隣の細胞に活動電位を伝播させるよりも弱い。
RRPは通常以上の刺激によってもたらされ、細胞の脱分極と活動電位の発生につながる。
「超正常期」とは、通常より弱い刺激で不整脈が発生する可能性のある過興奮状態をいい、心室細動を回避するために除細動中の同期が必要となる(図2)。 不応期。 ARP: Absolute Refractory Period(絶対不応期);ERP。 有効不応期、RRP: 相対的不応期、SNR:Supranormal Refractory Period
高カリウム血症の分類と原因
分類
高カリウム血症は5.5~6.0mmol/Lで軽度、6.1~6.9mmol/Lで中等度、7で重度と分類される。高カリウム血症は、代償機構が不均衡に対処できなくなったときに発生するため、通常は多因子性である。 食事による経口摂取、またはペニシリンGのようなカリウムを含む液体の静脈内投与。
高カリウム血症の影響
軽度の高カリウム血症は、倦怠感、筋力低下、知覚異常などの曖昧な症状のため、検査で偶然発見される無症状のことが多いです。 重症の高カリウム血症では、骨格筋の筋力低下や麻痺という形で神経筋機能に影響を与えるが、心毒性が支配的であり、その前段階であるため、頻度の高い症状ではない。 5352>
- 5.5mEq/L以上では、カリウムチャネルのコンダクタンスの増加によりlkr電流が増加し、表面心電図のピークT波の形で急速な再分極を引き起こす。 このT波は150-250msecの短い持続時間で心筋梗塞やCVAのT波と区別できる。
- カリウム濃度が6.5mEq/L以上では、閾値以下の脱分極が持続する状態となり、心房および心室の脱分極が遅延するようになる。 活動電位の第0相の減少により活動電位が長くなり、心室内伝導と房室伝導の遅延を生じる。 表面心電図では、P波の平坦化と消失、QRS複合の拡大が見られるようになる。 心室内伝導の遅延が大きくなると、表面心電図は左右の束縛ブロックの徴候を示すようになる。 高カリウム血症では、QRS複合体の初期や終末部分だけでなく、全体にわたって遅延が持続することで、束枝病と区別することができます
- 10mEq/Lでは、もはや洞房伝導は起こらず、加速した接合リズムが代用されます。 心室性不整脈は、広がったQRS複合体とT波が合流し、最終的に古典的な正弦波パターンを形成して発症する。 これが起こると、VFと不全収縮が起こり、心停止に至ります。
- 時には、カリウムの変化速度、カルシウム濃度、pH、ナトリウム濃度などの病因やその影響力の変動により、変化が不規則で予測できず、心電図は正常からアシストールにジャンプすることがある。 したがって、高カリウム血症は、カリウム値が6.5mmol/L以上になったとき、あるいはカリウム値に関係なく高カリウム血症の心電図所見があるときに、緊急に治療する必要があります。 急性高カリウム血症に関連するその他の報告としては、心電図記録上、筋細胞の再分極の狂いによる巨大なST-Tセグメント、短いPRおよびQT間隔、洞性頻脈、洞性徐脈、洞房リズム、第1度および第2度心ブロックがあり、疑似MIの像が描かれる。
代謝作用
高カリウム血症は、水素イオンと引き換えにカリウムの細胞内への取り込みを促進するため、高クロル性代謝性アシドーシスとなる。 これにより細胞内アルカローシスが生じ、近位尿細管での腎臓アンモニア産生が抑制され、尿中アンモニウムと酸の排泄が減少し、IV型腎尿細管アシドーシスとなる .
ナトリウムカリウムポンプ
ナトリウムカリウムATPアーゼは1957年にSkouによって発見され、後にその発見で1997年のノーベル化学賞の一部を授与されました。
ナトリウムカリウムポンプは、ATPの加水分解と、電気化学的勾配に逆らって2つのカリウムイオンと交換する3つのナトリウムイオンの細胞内輸送を結びつけることによって機能します。 ジギタリスやジゴキシンの分子標的であり、18世紀からジギタリスの抽出物として使用されている。
ナトリウムカリウムポンプの作用は、リン酸化タンパク質であるホスレマンによって制御されており、その非リン酸化はポンプの抑制につながり、リン酸化はポンプ活性の上昇につながる。 リン酸化部位は3箇所、パルミトイル化部位は2箇所、グルタチオニル化部位は1箇所であり、ポンプの活性化や阻害を行うことができるシグナルが多数存在することが説明できる
ナトリウムカリウム・ポンプ自体は複数のサブユニットと複数のアイソフォームからなる酵素である。 その機能には、αサブユニットとβサブユニット(主に心臓のB1)の存在が不可欠である。 最近、腎臓で第3のタンパク質γサブユニットが同定されたが、現在までのところその機能は不明である。
αサブユニットはナトリウムカリウムポンプ酵素の触媒的中核である。 約100kDaで、ナトリウム、カリウム、ATP、ウアバインのような強心性ステロイドの結合部位を含んでいる。 正常な心筋細胞ではα1とα2だけが有意に存在し、機能的にはナトリウム・カルシウム交換体(NCX)と結合している。 α3は実験的心不全モデルにおいてα2に取って代わることが報告されている。
最近の実験データは、興奮-収縮(E-C)カップリングの調節にポンプのα1α2サブユニットの両方が関与していることを支持している。 一方、E-Cカップリングの他の重要な構成要素とともにT-チューブに集中して発現しているα2は、主に収縮力に焦点を当てていると考えられている。 ATP、細胞内ナトリウム、小胞体下の障壁とファジースペース、膜電位、細胞内シグナル伝達経路(アドレナリンシグナル伝達経路、プロテインキナーゼA & C、一酸化窒素、ホスホールマン)、小分子による直接制御(脂質、内因性心筋ステロイド)、その他の関連タンパク質(カベオラとカベオリン、アンキリン)である。
結論
高カリウム血症は臨床的な課題であり、入院患者の最大10%に見られることがあります。 その最終結果は生命を脅かすものです。 体内のすべての細胞は最終的にナトリウム・カリウムポンプの影響を受けており、虚血した心筋はカリウムを細胞外に押し出し、不整脈の閾値を低下させることが知られており、心室性不整脈が低分極を悪化させ閾値をさらに低下させる可能性があるため、カリウム・ナトリウム酵素の制御が心停止の結果を良好に変え、現在のCPRガイドラインを書き換えることができるので、さらなる研究に焦点を当てる必要がある。