ABOVE: modificado de © ISTOCK.com, tera vector
Casi siempre, construir algo es más difícil que derribarlo. Del mismo modo, introducir genes supone un reto mayor que eliminarlos. Es una realidad que los investigadores tendrán que superar para sacar el máximo partido a la edición de genes. La introducción de genes permite a los científicos estudiar los efectos de variantes genéticas específicas, utilizar genes informadores como la proteína verde fluorescente para rastrear los productos genéticos en el tiempo y el espacio, investigar la regulación del genoma y, en última instancia, reparar los genes causantes de enfermedades. «Es una forma muy eficaz de interrogar cada base de un gen», afirma Greg Findlay, candidato a doctor en la Universidad de Washington.
CRISPR-Cas9, una tecnología de edición de genes conocida por su facilidad de uso, puede eliminar o introducir genes. La eliminación de un gen implica la inserción de CRISPR-Cas9 en una célula mediante un ARN guía que dirige la herramienta al gen de interés. Allí, Cas9 corta el gen, atravesando ambas cadenas de ADN, y el mecanismo de reparación del ADN de la célula arregla el corte mediante un proceso llamado unión de extremos no homólogos (NHEJ). La NHEJ es muy eficaz, pero inexacta. El proceso tiende a introducir errores en forma de pequeñas inserciones o deleciones que suelen ser suficientes para eliminar el gen.
Sin embargo, para eliminar un gen, los cortes deben repararse con mucha precisión, sin inserciones ni deleciones adicionales. Para ello es necesario aprovechar un segundo mecanismo de reparación del ADN llamado reparación dirigida por homología (HDR), que -al menos en las células de mamíferos- se produce con menos eficacia, por lo que su frecuencia es menor que la de la NHEJ. Para complicar aún más el proceso, algunos loci de genes y tipos de células son intrínsecamente menos hospitalarios para la edición de CRISPR-Cas9.
En los últimos años, los investigadores han desarrollado muchas estrategias nuevas para aumentar la eficiencia de la eliminación de genes grandes y pequeños mediante CRISPR-Cas9, y en el camino han propuesto y probado nuevas aplicaciones para este tipo de edición de genes. Aquí, The Scientist explora algunos de los enfoques más prometedores.
Selección
Investigador: Jon Chesnut, director senior de biología sintética R&D, Thermo Fisher Scientific
Proyecto: Al desarrollar un kit de etiquetado de genes llamado Truetag que Thermo Fisher pondrá en el mercado a finales de este año, Chesnut utilizó marcadores seleccionables para mejorar
la eficacia. Un marcador seleccionable -en este caso, un gen de resistencia a los antibióticos- se adhiere a una etiqueta de proteína fluorescente y se introduce en células de mamífero. A continuación, esas células se cultivan con el antibiótico asociado. El gen de resistencia confiere una ventaja selectiva a las células que lo portan; sólo ellas son capaces de crecer y, por tanto, las que crecen contienen la etiqueta genética de interés. Aunque la eficacia de la inserción del gen sea baja, los investigadores pueden utilizar la selección con antibióticos durante una semana o más para terminar con un alto porcentaje de células con inserciones exitosas.
Usando el antibiótico puromicina o blasticidina con el kit, el equipo de Chesnut consiguió aumentar la tasa de inserción del gen del 10 al 30% al 90% o más en algunas poblaciones celulares. Unos pocos genes especialmente difíciles pasaron de una tasa de inserción de menos del 1 por ciento a más del 90 por ciento. Es importante probar varias dosis de antibióticos en la línea celular que se planea utilizar para encontrar la dosis correcta, dice Chesnut: se quiere matar a las células sin inserciones pero no a las células con inserciones exitosas.
Pruébelo: Los marcadores seleccionables funcionan mejor cuando el gen de interés está muy expresado, dice Chesnut. «Si no lo está, es posible que aún se consiga la selección, pero puede que no se consiga una expresión suficiente de su etiqueta de proteína fluorescente para poder detectarla». Además, se aplican las limitaciones generales de CRISPR-Cas9. «Hay regiones del genoma que no se cortan muy bien con CRISPR, y todavía no sabemos por qué», añade. Además, algunos tipos de células no aceptan fácilmente el ADN, el ARN o los complejos ARN-proteína extraños, que son los tres métodos de administración de CRISPR-Cas9.
Para tener más suerte en la inserción de marcadores seleccionables, hay que asegurarse de que haya una secuencia llamada PAM, una etiqueta corta en el ADN diana que CRISPR-Cas9 debe reconocer antes de cortar, dentro de los 10 pares de bases del sitio de inserción del gen deseado, dice Chesnut. Si se aleja más del sitio de corte, la eficacia de la inserción puede ser demasiado baja para ser funcional. Sin un sitio PAM, se puede probar con TALENs o nucleasas de dedos de zinc, aunque esas técnicas de edición de genes más antiguas son más complicadas que CRISPR.
Inhibición de tiempo
Investigador: Jacob Corn, biólogo del genoma, Instituto Federal Suizo de Tecnología, Zúrich
Proyecto: Los investigadores no entienden por qué la vía NHEJ supera ampliamente a la vía HDR en las células de mamíferos. «Las levaduras hacen la HDR como un loco», dice Corn. En un esfuerzo por acelerar este proceso de reparación del ADN en las células humanas y mejorar el control del knock-in de los genes, él y su equipo están tratando de determinar cómo se regula la HDR. Examinaron las células humanas en busca de genes cuyo derribo condujera a un aumento de la HDR en la célula, y luego buscaron pequeñas moléculas inhibidoras de esos genes. Uno de los genes que aparecieron codifica el CDC7, una quinasa que regula la transición del ciclo celular a la fase S; su inhibidor, el XL413, multiplicó por dos o por tres la eficiencia del knock-in del gen (BioRXiv, DOI: 10.1101/500462, 2018). Esto se debe a que la HDR solo se produce en algunas partes del ciclo celular, incluida la fase S, afirma Corn. Si añades el inhibidor XL413 al mismo tiempo que utilizas CRISPR-Cas9 para editar tu gen objetivo, las células se acumulan en la fase inmediatamente anterior a la fase S. Luego se elimina XL413, y todas las células entran en la fase S y aumentan la eficiencia del knock-in.

Corn ha utilizado esta técnica en muchas líneas celulares humanas inmortalizadas y en células T humanas. Puede eliminar tramos cortos de ADN, como los SNP, así como grandes genes. No hay razón para que no funcione en ratones, dice, aunque no lo ha probado.
Prueba: «El momento es absolutamente clave», dice Corn. El Cas9 debe cortar el ADN al mismo tiempo que se añade el XL413. Si se inhibe primero y luego se libera mientras se edita con CRISPR-Cas9, la eficiencia de la recombinación homóloga se triplica en lugar de aumentar, porque las células se liberan en la fase incorrecta del ciclo celular.
Y como con cualquier esfuerzo de HDR, Corn dice, siempre ejecute un control sin nucleasas para asegurarse de que no está amplificando accidentalmente el ADN contaminante que está flotando alrededor de su laboratorio. Después de introducir el knock-in, «secuencia, secuencia, secuencia, secuencia», dice. El mero hecho de utilizar un sistema reportero, como una etiqueta de proteína fluorescente, para demostrar que la inserción del gen se ha realizado con éxito puede ser contraproducente. La secuenciación verifica que las inserciones se han realizado en el lugar correcto.
Jugar a largo plazo
Investigador: Channabasavaiah Gurumurthy, director del centro de ingeniería del genoma del ratón, Centro Médico de la Universidad de Nebraska
Proyecto: Hace unos años, reflexionando sobre la dificultad de introducir genes al intentar hacerlo en cigotos de ratón, Gurumurthy y sus colegas tuvieron una revelación.
Los investigadores estaban insertando con éxito ADN corto y monocatenario, así que ¿por qué no intentar hacer un knock-in insertando ADN largo y monocatenario? De hecho, el enfoque, que Gurumurthy llama Easi-CRISPR (adiciones eficientes con inserciones de ssDNA -CRISPR), aumenta la eficiencia en 2,5 veces, y el uso de ADN monocatenario reduce la tasa de inserciones fuera del objetivo 100 veces en el cultivo celular (Nat Protoc 13:195-215, 2018; Nature 559:405-09, 2018). «Es bastante enorme», dice. En el laboratorio de Gurumurthy, Easi-CRISPR ha generado una línea de ratón knock-in para 9 de cada 10 genes que han probado. Un colaborador también lo ha utilizado en células T humanas para crear células CAR-T, células inmunitarias específicas para pacientes que luchan contra el cáncer.
Pruébelo: Gurumurthy advierte que Easi-CRISPR está lejos de ser infalible. A veces la técnica inserta sólo una parte del gen. Además, añade, puede desordenar los brazos de homología, es decir, las secuencias cortas a ambos lados del gen que lo dirigen a su objetivo correcto en el genoma. Y algunos loci son inexplicablemente más difíciles de insertar que otros.
Pocos proveedores comerciales diseñan y sintetizan ADN largo y monocatenario a medida. Se puede hacer el propio, pero la estabilidad del ADN monocatenario varía; las secuencias menos estables tendrán un menor rendimiento, por lo que es posible que haya que sintetizar más, dice Gurumurthy.
Los investigadores que no puedan insertar CRISPR en embriones de ratón unicelulares pueden pagar a una instalación central para que haga los ratones con su secuencia de ADN, dice Gurumurthy. Las instalaciones centrales como la suya cobran entre 5.000 y 15.000 dólares por generar una o dos parejas reproductoras; las instalaciones comerciales cobran entre 20.000 y 50.000 dólares, dice.
Knock-in By Numbers
Investigador: Greg Findlay, candidato a doctor en el laboratorio de Jay Shendure, Universidad de Washington
Proyecto: Findlay y sus colegas pretendían mejorar la forma en que los médicos interpretan las mutaciones en el gen del cáncer de mama y ovario BRCA1. Ese gen tiene miles de variantes, pero los investigadores desconocen cómo afectan la mayoría de ellas a su función. Para estudiar el impacto de estas variantes, utilizaron una técnica de knock-in que desarrollaron llamada edición del genoma por saturación (Nature, 562:217-22, 2018).
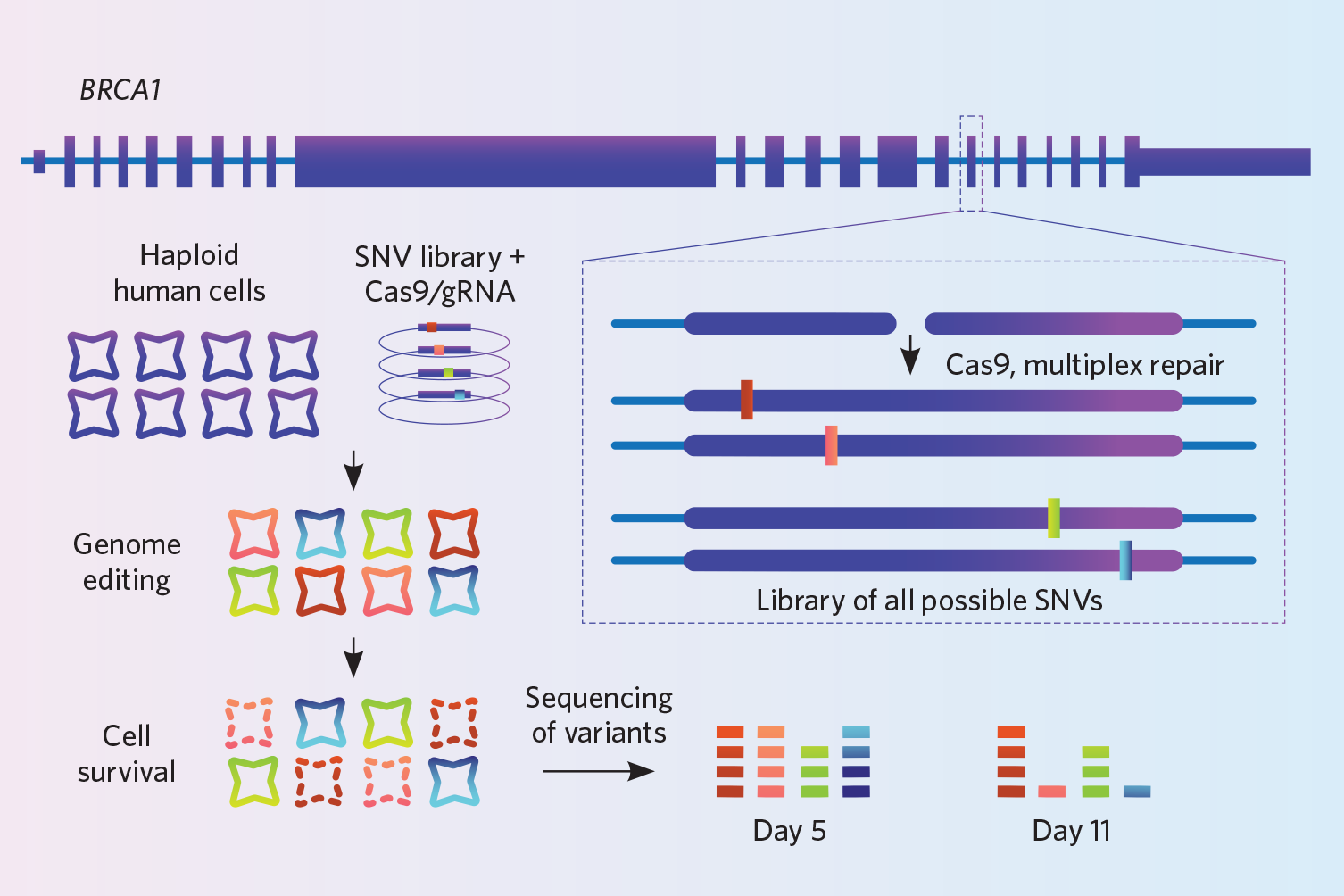
En una línea celular humana haploide inmortalizada, utilizaron CRISPR-Cas9 para eliminar 4.000 variantes diminutas en millones de células a la vez in vitro. El genoma se corta en el mismo punto en cada célula, pero el genoma de cada célula recibe una variante diferente. Para promover la HDR, también eliminaron el gen de la ligasa4, desactivando la vía de reparación NHEJ, un paso que triplicó la eficiencia, dice Findlay. Por último, dado que todos los knock-ins de las células son diferentes, secuenciaron las células en profundidad, cubriendo la misma región genómica millones de veces, para asegurarse de que realmente habían eliminado las 4.000 variantes que querían estudiar. Secuenciaron en dos momentos y dedujeron que los knock-ins que no aparecían en la secuenciación del segundo momento eran los que interferían en la función del gen, porque las células que los portaban debían haber muerto.
Prueba: El equipo de Findlay hizo fabricar para ellos los oligos de ADN de las 4.000 variantes en un microarray. Se pueden comprar matrices de entre 6.000 y 250.000 oligos, por lo que hay que considerar la posibilidad de sacar más provecho combinando varios experimentos en la misma matriz, dice Findlay. Su laboratorio paga unos 5.000 dólares por 100.000 oligos.
Esta estrategia tiene sus limitaciones: hasta ahora sólo se ha utilizado para eliminar variantes de un solo nucleótido, y todas las ediciones tienen que estar en el mismo gen. El método funciona mejor cuando se edita una región de ADN bastante estrecha, de unos 110-120 pares de bases, porque los oligos de ADN más largos tendrían demasiados errores, dice Findlay. También es importante secuenciar muy profundamente para asegurarse de que se tiene en cuenta todo el número de variantes que se pretende eliminar.