- Una revisión clinicopatológica
- INTRODUCCIÓN
- Distrofias corneales epiteliales-estromales
- DISTROFIA CORNEAL DE REIS-BUCKLERS
- Distrofia corneal en retícula
- Distrofia corneal granular, TIPO I
- Distrofia corneal granular, tipo II
- Distrofia corneal macular
- Distrofia corneal macular
- Distrofia corneal de Schnyder (SCD)
- Tabla 4: Mnemotecnia para recordar las distrofias del estroma corneal
- VISIÓN: DISTROFIAS CORNEALES
- EPIDEMIOLOGÍA
- SIGNOS
- SINTOMAS
- Tratamiento
Una revisión clinicopatológica
Emily S. Birkholz, MD, Nasreen A. Syed, MD, y Michael D. Wagoner, MD, PhD
El 17 de agosto de 2009
Revisión mayor: Chaunhi Van, MD and Nasreen Syed, MD
August 20, 2015
INTRODUCCIÓN
Las distrofias epitelio-estromales y estromales de la córnea son un grupo de trastornos hereditarios de la córnea que están causados por la acumulación progresiva de depósitos dentro de las capas de la córnea. Estos depósitos no están causados por una inflamación, una infección o un traumatismo, sino por mutaciones genéticas que conducen a la transcripción de proteínas aberrantes que dan lugar a la acumulación de material insoluble dentro de la córnea. Los trastornos pueden afectar o no a la visión y pueden ser simétricos o no (1). El sistema de clasificación del Comité Internacional para la Clasificación de las Distrofias Corneales (IC3D) de 2015 ha dividido las distrofias corneales en 4 categorías: distrofias epiteliales y subepiteliales, distrofias epiteliales-estromales, distrofias estromales y distrofias endoteliales. La mayoría de las distrofias que antes se consideraban estromales se clasifican ahora como distrofias epiteliales-estromales o distrofias estromales. En las tablas 1 y 2 se enumeran las distrofias epitelio-estromales y las distrofias estromales (2). La antigua clasificación de las distrofias estromales de la córnea figura en la tabla 3.
- Distrofia corneal de Reis-Bucklers
- Distrofia corneal de Thiel-Behnke
- Distrofia corneal reticular, tipo 1 y variantes
- Distrofia corneal granular, tipo 1
- Distrofia corneal granular, tipo 2
- Distrofia corneal macular
- Distrofia corneal de Schnyder
- Distrofia corneal estromal congénita
- Distrofia corneal amorfa posterior
- Distrofia nubosa central de Francois
- Pre.Distrofia corneal de Descemet
Tabla 3. Clasificación antigua de las distrofias del estroma corneal
- Distrofia corneal reticular
- Distrofia corneal granular
- Distrofia corneal de Avellino
- Distrofia corneal macular
- Distrofia gelatinosa tipo gota
- Distrofia corneal de Schnyder
- Distrofia Francois-Neetans Fleck
- Distrofia estromal hereditaria congénita
.distrofia gelatinosa
Distrofias corneales epiteliales-estromales
Las distrofias epiteliales-estromales están causadas por mutaciones en el gen inducido por el factor de crecimiento transformante beta (TGFβI), también conocido como el gen BIGH3. El TGFβI está localizado en el cromosoma 5q31 y codifica la queratoepitelina, una proteína secretada por el epitelio corneal. Esta proteína actúa como una proteína de adhesión y está presente en el estroma normal. Al ser una proteína pequeña, aproximadamente del tamaño de la albúmina, tiene la capacidad de difundirse a través del estroma corneal. Cuando se produce una mutación en el gen del TGFβI, la estructura de la queratoepitelina es anormal y se produce una acumulación de la proteína insoluble o de sus fragmentos proteolíticos en la córnea (1, 3). Curiosamente, la mutación del gen TGFβI se descubrió en parte en la Universidad de Iowa. Un grupo de investigadores y clínicos, entre los que se encontraban Edwin M. Stone, Robert Folberg y Jay H. Krachmer, mapearon la distrofia granular tipo I, la distrofia granular tipo II y la distrofia reticular en el cromosoma 5q en 1994 (4). Hasta la fecha, se han identificado 63 mutaciones diferentes en el gen TGFβI. No se han identificado tratamientos eficaces para prevenir o atenuar el depósito de la queratoepitelina. Las distrofias suelen tener una herencia autosómica dominante y afectan a la capa de Bowman y al estroma (3).
DISTROFIA CORNEAL DE REIS-BUCKLERS
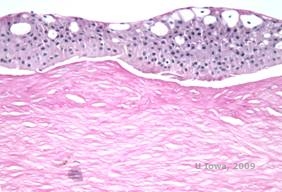
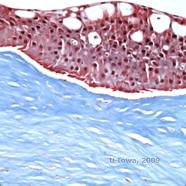
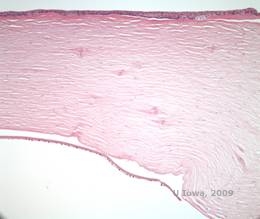
Reis-Bücklers, antes conocida como distrofia corneal granular de tipo III o distrofia corneal de Bowman de tipo I, suele presentar córneas normales al nacer, pero desarrolla erosiones recurrentes dolorosas, opacificación y pérdida de visión progresiva en la primera década de vida (1). En la capa de Bowman y en el estroma anterior se localizan opacidades irregulares, de color blanco-grisáceo y de aspecto geográfico. En fases más avanzadas de la enfermedad, las opacidades pueden extenderse al limbo y al estroma más profundo (2). La histopatología revela depósitos estromales y subepiteliales anteriores de material hialino que interrumpen y a menudo sustituyen la capa de Bowman (véanse las figuras 1A y 1B). Los depósitos se tiñen de rojo con la tinción de tricrómico de Masson (2). El material hialino consiste en cuerpos similares a varillas desde el punto de vista ultraestructural, lo que ayuda a distinguirla de la distrofia corneal de Thiel-Behnke (1, 2).
| A: H&E de Reis Bückler que muestra la destrucción de la capa de Bowman y el epitelio irregular | B. Tinción de tricrómico de Masson que demuestra la tinción epitelial |
 |
 |
Distrofia corneal en retícula
La distrofia corneal en retícula (LCD) es la más común de las distrofias epitelio-estromales de la córnea. Se trata de una enfermedad bilateral autosómica dominante que suele presentarse hacia el final de la primera década de vida con síntomas de erosiones corneales recurrentes y disminución de la visión. Se caracteriza por la presencia de líneas de celosía, que son opacidades refractarias lineales, orientadas radialmente y ramificadas, descritas como «de cristal», localizadas en el estroma anterior (véanse las figuras 2A y 2B). Estas líneas reticulares se encuentran inicialmente en la córnea central superficial. A medida que la enfermedad progresa, se extienden más profundamente y periféricamente en el estroma, evitando el limbo (1, 2). Otros hallazgos del examen incluyen opacidades similares a motas, puntos blancos subepiteliales y opacidad estromal «en vidrio deslustrado», que comienza en el centro y se vuelve más difusa (2). Muchos pacientes con LCD requerirán una intervención quirúrgica para tratar las erosiones recurrentes y la disminución de la visión. Si la enfermedad se localiza en la parte anterior del estroma, los pacientes a menudo pueden ser tratados con éxito mediante queratectomía fototerapéutica (PTK). Algunos requieren un trasplante de córnea. Dado que la queratoepitelina, la proteína producida por el gen TGFβI, se produce principalmente en el epitelio corneal, la enfermedad tiende a reaparecer en los injertos de córnea (1).
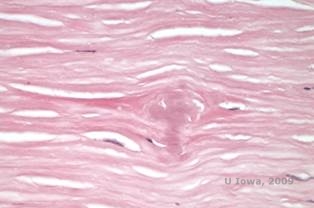
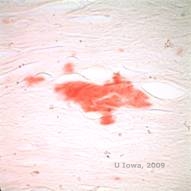
En la LCD, los depósitos amiloides se acumulan entre la membrana basal epitelial y la capa de Bowman, así como en el estroma, causando una distorsión de la arquitectura laminar. Los depósitos se tiñen positivamente con la inmunohistoquímica utilizando anticuerpos contra la queratoepitelina (2). Los depósitos aparecen como depósitos amorfos de color rosa en la tinción de hematoxilina y eosina (H&E) (Ver Figura 1C y 1D) y se tiñen con la tinción de rojo Congo demostrando la clásica birrefringencia verde manzana en la polarización cruzada (Ver Figura 2E y 2F) (1). La ausencia o el adelgazamiento de la capa de Bowman, la atrofia epitelial y la degeneración epitelial basal también pueden encontrarse en la histopatología de la LCD (2).
La LCD tipo I es la forma clásica de LCD causada por una mutación en el gen TGFβI que da lugar a un depósito amiloide aislado en la córnea. Se han identificado cuatro variantes de LCD: LCD tipo IIIA, tipo I/IIIA, tipo IV y amiloidosis polimórfica. Las variantes de la LCD se presentan más tarde en la vida que la LCD clásica. La LCD tipo IIIA se presenta en la 5ª-7ª década, normalmente con erosiones epiteliales. Presenta líneas reticulares más gruesas, descritas como de «aspecto ropy», que se extienden hasta el limbo. La LCD de tipo I/IIIA tiene líneas de rejilla finas. La LCD tipo IV se presenta en la 7ª-9ª década con pequeñas líneas reticulares. Los depósitos amiloides en la LCD tipo IV se encuentran en el estroma profundo y rara vez se producen erosiones epiteliales. Las líneas reticulares están ausentes en la amiloidosis de tipo polimórfico y rara vez se producen erosiones epiteliales (2).
La LCD tipo II es un síndrome de amiloidosis sistémica conocido como síndrome de Meretoja que afecta a la piel, los nervios craneales y la córnea. Se presenta en la edad adulta temprana con neuropatías periféricas, neuropatías craneales, facies de sabueso, piel seca, blefarochalasis, labios protuberantes y líneas de entramado corneal. Este tipo se ha relacionado con el gen de la gelsolina en el cromosoma 9, que codifica para una proteína precursora de amiloide que funciona para eliminar la actina de los sitios de lesión e inflamación (1). El nombre es erróneo y no se considera una variante de la distrofia corneal reticular (2).
| A: Ojo izquierdo en retroiluminación que demuestra depósitos estromales anteriores en la distrofia corneal en celosía | B: Ojo izquierdo con mayor potencia que muestra depósitos estromales anteriores lineales. |
 |
 |
| C: Tinción H&E de la córnea con retículo. Obsérvense los depósitos amorfos de color rosa en el estroma | D: Una vista más cercana de los depósitos amorfos de color rosa |
 |
 |
| E: Tinción de rojo Congo, resaltando el amiloide | F: birrefringencia verde manzana del amiloide con polarización cruzada. |
 |
 |
Distrofia corneal granular, TIPO I
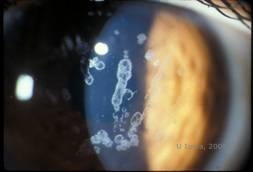
Distrofia corneal granular, tipo I (GCD1) es una enfermedad bilateral, autosómica dominante, asociada a una mutación en el gen TGFβI que conduce a la deposición de un material hialino en el estroma corneal. Suele presentarse al principio de la primera década de vida con opacidades de color blanco-grisáceo, similares a migas, en el estroma anterior y medio, que se extienden al estroma posterior en la enfermedad avanzada (1, 2). Estas opacidades son depósitos discretos localizados en el centro, con córnea clara localizada en la periferia y córnea clara entre los depósitos (Ver Figura 3A y 3B). La enfermedad suele ser asintomática al principio, pero con el tiempo las opacidades pueden unirse y provocar una disminución de la visión. En la DGC pueden producirse erosiones corneales recurrentes, pero con una incidencia menor que en la LCD (1, 5). Los pacientes también pueden experimentar deslumbramiento y fotofobia (2). El tratamiento al principio del proceso de la enfermedad suele ser sólo de observación. Sin embargo, a medida que la enfermedad progresa, puede ser necesaria la PTK y el trasplante de córnea para mejorar la visión y los síntomas de erosión. Al igual que la LCD, la enfermedad puede reaparecer en los injertos de córnea.
Desde el punto de vista histopatológico, las opacidades son depósitos eosinófilos que a menudo se describen como «similares a caramelos de roca» en el estroma anterior, hechos de un material similar a la hialina. Con el tiempo, los depósitos progresan hacia el estroma corneal más profundo. El material hialino se tiñe de rojo brillante con la tinción de tricrómico de Masson (véanse las figuras 3C y 3D).
| A:Fotografía con lámpara de hendidura de la distrofia corneal granular, tipo I | B: Obsérvense los depósitos estromales «en forma de migas» con un estroma intermedio claro. |
 |
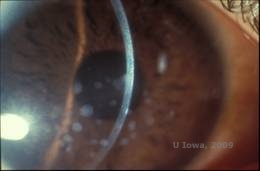 |
| C: Tinción H& E de la córnea que muestra depósitos hialinos eosinofílicos «como caramelos de roca» en el estroma | D: El material hialino se tiñe de rojo brillante con Masson-Trichrome |
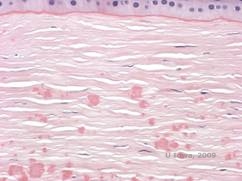 |
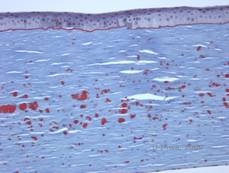 |
Distrofia corneal granular, tipo II
Distrofia corneal granular, tipo II (GCD2), anteriormente conocida como distrofia corneal de Avellino o combinada granular-latinosa, es una enfermedad autosómica dominante vinculada a una mutación en el gen TGFβI que conduce a una deposición tanto hialina como amiloide en el estroma corneal. Normalmente, los pacientes se presentan en la segunda década de vida con pequeños puntos blanco-grisáceos en el estroma superficial. Las opacidades también pueden tener una forma espinosa, anular o estrellada. En la retroiluminación, son parcialmente translúcidas. Más adelante en el proceso de la enfermedad, pueden desarrollar también líneas reticulares (véase la figura 4A y 4B). Estas líneas no se cruzan entre sí y parecen más blancas y menos refractarias que las líneas reticulares. Los síntomas de la GCD2 son dolor con erosiones epiteliales y deterioro visual (2).
Histopatológicamente, la córnea tendrá depósitos estromales que se tiñen de rojo con el Tricrómico de Masson, indicando la presencia de hialina (Ver Figura 4C). Además, la tinción con rojo Congo mostrará una birrefringencia verde manzana en la polarización cruzada que indica la presencia de amiloide (véase la figura 4D). Se creía que la enfermedad tenía su origen en una familia de Avellino, Italia. Sin embargo, en la actualidad se ha informado de la existencia de la DGC tipo II en pacientes de muchos otros países (2,5), siendo la mayor prevalencia en Asia oriental.
| A: Distrofia de Avellino que muestra depósitos de tipo reticular y granular en el estroma corneal | B. Tinción de tricrómico de Masson que muestra depósitos hialinos en el estroma anterior |
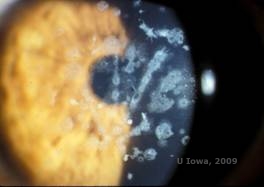 |
 |
| C: Tinción de rojo Congo que muestra depósitos amiloides rosados y amorfos en el mismo espécimen corneal. | D: La polarización cruzada revela una birrefringencia verde manzana que indica amiloide. |
 |
 |
Distrofia corneal macular
Distrofia corneal macular
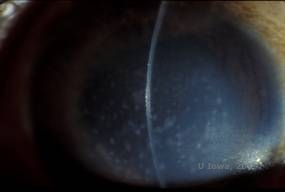
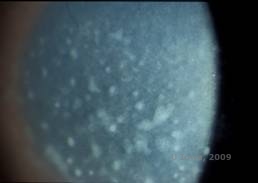
La distrofia corneal macular (MCD) es una enfermedad autosómica recesiva causada por una mutación en el gen de la carbohidrato sulfotransferasa 6 (CHST6) en el cromosoma 16 que conduce a un defecto en la síntesis de queratán sulfato el principal glicosaminoglicano de la córnea. Es menos frecuente que la LCD o la DGC, pero tiende a afectar más gravemente a la visión. Aunque la DMC es menos común en todo el mundo que la LCD o la DGC, es la más común de las distrofias del estroma corneal en lugares como Islandia y Arabia Saudí (2,6). Las lesiones estromales anteriores de color blanco grisáceo, similares a las de la DGC1, aparecen en la córnea en la primera década de vida. Sin embargo, a diferencia de la GCD1, hay una neblina estromal entre los depósitos y toda la córnea, de limbo a limbo, suele estar implicada (véanse las figuras 5A y 5B). La córnea es delgada y, a medida que el trastorno progresa, la membrana de Descemet se vuelve gris y desarrolla guta. Pueden producirse erosiones epiteliales, pero menos en la MCD que en la LCD. Los pacientes suelen desarrollar una pérdida visual grave entre la segunda y la tercera década de vida debido a la opacidad corneal difusa. La PTK puede realizarse en algunos casos tempranos de MCD. Sin embargo, esta afección no suele ser tan susceptible a la PTK como la distrofia reticular o granular y a menudo requiere un trasplante de córnea para su tratamiento (7). La recurrencia en los injertos es menos frecuente en la DMC que en la distrofia granular o lattice (1,2,5,6,8).
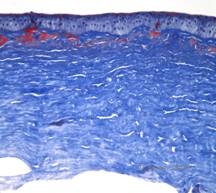
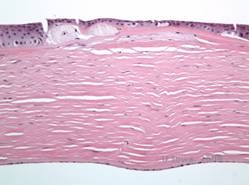
Los depósitos estromales en la DMC se componen de mucopolisacáridos que se acumulan dentro del retículo endoplásmico de los queratocitos del estroma corneal, extracelularmente entre las láminas estromales, y dentro del epitelio, la membrana de Descemet y el endotelio. Estos depósitos se tiñen de azul con el azul Alcian (véase la figura 5C y 5D) (1). Hay roturas en la capa de Bowman y en las gutañas con engrosamiento de la membrana de Descemet (2).
Se han descrito tres subtipos de MCD basados en la presencia o ausencia de queratán sulfato inmunoreactivo en varios tejidos. El tipo I no tiene queratán sulfato inmunorreactivo en el estroma corneal, los queratocitos, los sueros o el cartílago, y es la variante más común de la MCD en todo el mundo. El tipo IA carece de queratán sulfato en el estroma, los sueros y el cartílago, pero tiene niveles detectables dentro de los queratocitos. El tipo II tiene sulfato de queratán presente en niveles muy reducidos en el estroma, los queratocitos, el suero y el cartílago (6).
| A: Fotografía con lámpara de hendidura de la distrofia corneal macular. | B: Nótese la neblina entre los depósitos del estroma corneal |
 |
 |
| C: H&E de córnea con distrofia macular. Obsérvense los depósitos del estroma anterior y la alteración de la capa de Bowman | D: Depósitos de mucopolisacáridos dentro de los queratocitos resaltados con la tinción de azul Alcian |
 |
 |
Distrofia corneal de Schnyder (SCD)
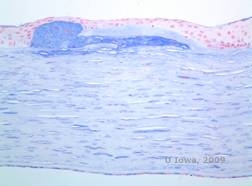
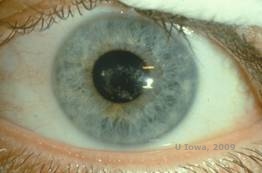
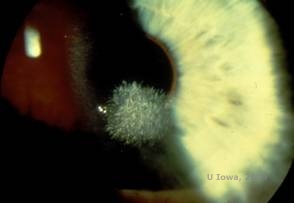
Distrofia corneal de Schnyder (SCD), anteriormente conocida como distrofia corneal cristalina de Schnyder, es una distrofia estromal corneal bilateral, autosómica dominante, vinculada a una mutación genética en el gen UbiA preniltransferasa que contiene el dominio 1 (UBIAD1) en el cromosoma 1. El defecto metabólico resultante de los queratocitos de la córnea conduce a la deposición de colesterol cristalino en el estroma. Sin embargo, la presencia de cristales no es absolutamente necesaria para el diagnóstico de la ECS. De hecho, sólo el 54% de los pacientes con ECF tienen cristales en la córnea. Normalmente, los pacientes se presentan en la segunda o tercera década con una opacidad corneal central en forma de anillo, con o sin cristales subepiteliales en forma de coma (véase la figura 6A y 6B). A continuación, el arcus lipoides aparece entre los 23 y los 38 años. Después de los 38 años, la opacidad progresiva de la córnea da lugar a una neblina panestromal que llega a la periferia media. La mayoría de los pacientes de más de 50 años presentan pérdida de visión fotópica, deslumbramiento y disminución de la sensibilidad corneal, por lo que pueden requerir un tratamiento quirúrgico que incluya el trasplante de córnea o la PTK. Pueden producirse recidivas en el injerto. La enfermedad se ha relacionado con la hipercolesterolemia, la hiperlipidemia y el genu valgum en algunos pacientes (2,5,9,10).
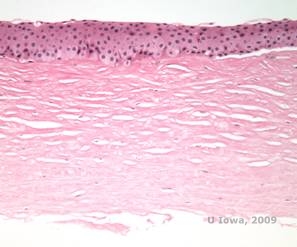
Histopatológicamente, los cristales de colesterol birrefringentes compuestos por fosfolípidos y colesterol se depositan dentro de las células epiteliales basales, los queratocitos, la capa de Bowman y entre las láminas estromales. Los lípidos se disuelven en el procesamiento histológico normal, por lo que deben obtenerse secciones congeladas de la córnea para demostrar la presencia de lípidos con las tinciones Oil-Red-O o Sudan black.
| A: Fotografía con lámpara de hendidura de la distrofia corneal de Schnyder. | B. Depósitos cristalinos localizados en el centro |
 |
 |
| C: H&E de córnea con DCCS | D. La tinción Oil Red O resalta los cristales de colesterol que aparecen de color rojo. |
 |
 |
La tabla 4 proporciona una mnemotecnia común para memorizar algunas de las distrofias corneales que afectan al estroma, la composición de su depósito, y se enumera el método de tinción de estos depósitos.
Tabla 4: Mnemotecnia para recordar las distrofias del estroma corneal
- Distrofia Marilyn-Macular
- Monroe-Mucopolisacárido
- Tinción siempre con azul alciano
- Gets-Granular Distrofia
- Her-Hyaline
- Tinción Man in-Masson Trichrome
- Distrofia Los-Lattice
- Angeles-Amyloid
- California-Congo Red
VISIÓN: DISTROFIAS CORNEALES
EPIDEMIOLOGÍA
|
SIGNOS
|
SINTOMAS
|
Tratamiento
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Conjunctival and Corneal Pathology. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3rd edition. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D classification of corneal dystrophies–edition 2. Cornea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. Clinical and genetic aspects of the TGFBI-associated corneal dystrophies. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nature genet. 1994; 6: 47-51.
- Bron AJ. Genética de las distrofias corneales: Lo que hemos aprendido en los últimos veinticinco años. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating Keratoplasty for Macular Corneal Dystrophy. Ophthalmology 2005;112: 220-224.
- Badr IA, Wagoner MD. Phototherapeutic Keratectomy for Macular Corneal Dystrophy. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetics of anterior and stromal corneal dystrophies. Seminars in Ophthalmology, 2008; 23: 1,9-17.
- Weiss JS. Distrofia corneal de Schnyder. Curr Opin Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. The gene for Schnyder’s crystalline corneal dystrophy maps to human chromosome 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Formato de citación sugerido: Van C, Syed NA. Epithelial-Stromal and Stromal Corneal Dystrophies: A Clinicopathologic Review. Revisión de ; EyeRounds.org. 20 de agosto de 2015. Disponible en: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm