- Eine klinisch-pathologische Übersicht
- EINLEITUNG
- Epithelial-stromale Hornhautdystrophien
- REIS-BUCKLERS CORNEAL DYSTROPHY
- Gitterhornhautdystrophie
- GRANULÄRE CORNEALE DYSTROPHIE, TYP I
- GRANULÄRE KORNEALDYSTROPHIE, TYP II
- STROMALE KORNEALE DYSTROPHIEN
- MAKULARE KORNEALE DYSTROPHIE
- SCHNYDER CORNEAL DYSTROPHY (SCD)
- Tabelle 4: Eselsbrücke zur Erinnerung an Hornhautstroma-Dystrophien
- ÜBERSICHT: CORNEAL STROMAL DYSTROPHIEN
- EPIDEMIOLOGIE
- SIGNS
- SYMPTOME
- Behandlung
Eine klinisch-pathologische Übersicht
Emily S. Birkholz, MD, Nasreen A. Syed, MD, und Michael D. Wagoner, MD, PhD
August 17, 2009
Hauptüberarbeitung: Chaunhi Van, MD und Nasreen Syed, MD
August 20, 2015
EINLEITUNG
Hornhaut-Epithel-Stroma- und Stroma-Dystrophien sind eine Gruppe von erblichen Erkrankungen der Hornhaut, die durch eine fortschreitende Ansammlung von Ablagerungen in den Schichten der Hornhaut verursacht werden. Diese Ablagerungen werden nicht durch Entzündungen, Infektionen oder Traumata verursacht, sondern durch genetische Mutationen, die zur Transkription abnormaler Proteine führen, was wiederum die Ansammlung von unlöslichem Material in der Hornhaut zur Folge hat. Die Störungen können das Sehvermögen beeinträchtigen oder nicht und können symmetrisch oder unsymmetrisch sein (1). Das Klassifizierungssystem des Internationalen Komitees für die Klassifizierung von Hornhautdystrophien (IC3D) aus dem Jahr 2015 hat Hornhautdystrophien in vier Kategorien eingeteilt: epitheliale und subepitheliale Dystrophien, epithelial-stromale Dystrophien, stromale Dystrophien und endotheliale Dystrophien. Die meisten Dystrophien, die früher als stromale Dystrophien galten, werden jetzt entweder als epithelial-stromale Dystrophien oder als stromale Dystrophien klassifiziert. In Tabelle 1 und 2 sind die epithelial-stromalen Dystrophien und stromalen Dystrophien aufgeführt (2). Die alte Klassifikation für stromale Hornhautdystrophien ist in Tabelle 3 aufgeführt.
- Reis-Bucklers Hornhautdystrophie
- Thiel-Behnke-Hornhautdystrophie
- Gitter-Hornhautdystrophie, Typ 1 und Varianten
- Granuläre Hornhautdystrophie, Typ 1
- Granuläre Hornhautdystrophie, Typ 2
- Makuläre Hornhautdystrophie
- Schnyder-Hornhautdystrophie
- Kongenitale stromale Hornhautdystrophie
- Posteriore amorphe Hornhautdystrophie
- Zentrale trübe Dystrophie von Francois
- Prä-Descemet-Hornhautdystrophie
Tabelle 3. Alte Klassifikation der stromalen Hornhautdystrophien
- Gitterförmige Hornhautdystrophie
- Granuläre Hornhautdystrophie
- Avellino-Hornhautdystrophie
- Makuläre Hornhautdystrophie
- Gelatinöse Tropfen-wie Dystrophie
- Schnyder-Hornhautdystrophie
- Francois-Neetans Fleck-Dystrophie
- Kongenitale hereditäre Stromadystrophie
Epithelial-stromale Hornhautdystrophien
Epithelial-stromale Dystrophien werden durch Mutationen im TGFβI-Gen (Transforming Growth Factor beta-induced) verursacht, auch bekannt als das BIGH3-Gen. TGFβI befindet sich auf Chromosom 5q31 und kodiert für Keratoepithelin, ein Protein, das vom Hornhautepithel sezerniert wird. Dieses Protein wirkt als Adhäsionsprotein und ist im normalen Stroma vorhanden. Da es ein kleines Protein von der Größe von Albumin ist, hat es die Fähigkeit, durch das Hornhautstroma zu diffundieren. Wenn eine Mutation im TGFβI-Gen auftritt, ist die Keratoepithelin-Struktur abnormal und es kommt zu einer Akkumulation des unlöslichen Proteins oder seiner proteolytischen Fragmente in der Hornhaut (1, 3). Interessanterweise wurde die TGFβI-Genmutation zum Teil an der Universität von Iowa entdeckt. Eine Gruppe von Forschern und Klinikern, darunter Edwin M. Stone, Robert Folberg und Jay H. Krachmer, ordnete 1994 die granuläre Typ I, granuläre Typ II und Gitterdystrophie dem Chromosom 5q zu (4). Bis heute sind 63 verschiedene Mutationen im TGFβI-Gen identifiziert worden. Es wurden noch keine wirksamen Behandlungen zur Verhinderung oder Abschwächung der Keratoepithelin-Ablagerung gefunden. Die Dystrophien haben typischerweise einen autosomal-dominanten Erbgang und betreffen die Bowman-Schicht und das Stroma (3).
REIS-BUCKLERS CORNEAL DYSTROPHY
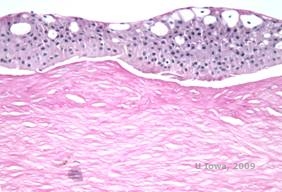
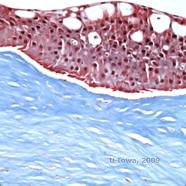
Reis-Bücklers, früher bekannt als Granuläre Hornhautdystrophie Typ III oder Corneal Dystrophy of Bowman’s type I, zeigen bei der Geburt typischerweise normale Hornhäute, entwickeln aber innerhalb des ersten Lebensjahrzehnts schmerzhafte, wiederkehrende Erosionen, Trübungen und fortschreitenden Sehverlust (1). Unregelmäßige, grau-weiße, geografisch anmutende Trübungen befinden sich in der Bowman-Schicht und im anterioren Stroma. In fortgeschrittenen Krankheitsstadien können sich die Trübungen auf den Limbus und das tiefere Stroma ausdehnen (2). Die Histopathologie zeigt anteriore stromale und subepitheliale Ablagerungen aus hyalinartigem Material, die die Bowman-Schicht unterbrechen und oft ersetzen (siehe Abbildung 1A und 1B). Die Ablagerungen färben sich mit der Masson-Trichrom-Färbung rot (2). Das hyalinartige Material besteht ultrastrukturell aus stäbchenförmigen Körpern, was zur Unterscheidung von der Thiel-Behnke-Hornhautdystrophie beiträgt (1, 2).
| A: H&E von Reis Bückler mit Zerstörung der Bowman-Schicht und unregelmäßigem Epithel | B. Masson-Trichrom-Färbung mit Epithelfärbung |
 |
 |
Gitterhornhautdystrophie
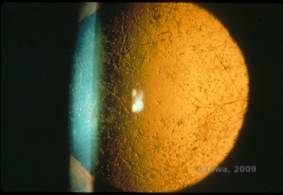
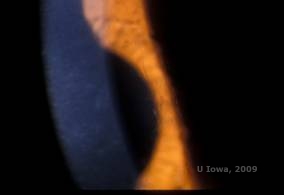
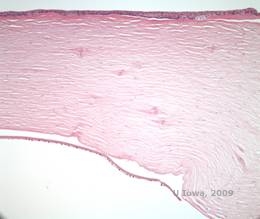
Die Gitterhornhautdystrophie (LCD) ist die häufigste der epithelial-stromalen Hornhautdystrophien. Es handelt sich um eine autosomal dominante, bilaterale Erkrankung, die typischerweise gegen Ende des ersten Lebensjahrzehnts auftritt und Symptome wie wiederkehrende Hornhauterosionen und verminderte Sehkraft aufweist. Sie ist gekennzeichnet durch Gitterlinien, lineare, radial orientierte, verzweigte refraktive Trübungen, die als „glasartig“ beschrieben werden und sich im vorderen Stroma befinden (siehe Abbildung 2A und 2B). Diese Gitterlinien finden sich zunächst in der oberflächlichen zentralen Hornhaut. Mit dem Fortschreiten der Erkrankung breiten sie sich tiefer und peripher im Stroma aus, wobei der Limbus ausgespart wird (1, 2). Zu den weiteren Untersuchungsergebnissen gehören fleckartige Trübungen, subepitheliale weiße Punkte und eine „glasige“ Stromatrübung, die zentral beginnt und dann immer diffuser wird (2). Bei vielen Patienten mit LCD ist ein chirurgischer Eingriff erforderlich, um wiederkehrende Erosionen und Sehschwächen zu behandeln. Wenn die Erkrankung anterior im Stroma lokalisiert ist, können die Patienten oft erfolgreich mit einer phototherapeutischen Keratektomie (PTK) behandelt werden. Einige benötigen eine Hornhauttransplantation. Da Keratoepithelin, das vom TGFβI-Gen produzierte Protein, hauptsächlich im Hornhautepithel gebildet wird, neigt die Krankheit dazu, in Hornhauttransplantaten zu rezidivieren (1).
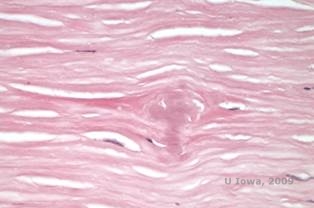
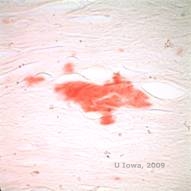
Bei LCD sammeln sich Amyloidablagerungen zwischen der epithelialen Basalmembran und der Bowman-Schicht sowie im Stroma an und verursachen eine Verzerrung der lamellaren Architektur. Die Ablagerungen werden in der Immunhistochemie mit Antikörpern gegen Keratoepithelin positiv angefärbt (2). Die Ablagerungen erscheinen als amorphe, rosafarbene Ablagerungen in der Hämatoxylin- und Eosinfärbung (H&E) (siehe Abbildung 1C und 1D) und zeigen in der Kongorotfärbung die klassische apfelgrüne Doppelbrechung bei Kreuzpolarisation (siehe Abbildung 2E und 2F) (1). Das Fehlen oder die Ausdünnung der Bowman-Schicht, eine Epithelatrophie und eine basale Epitheldegeneration können bei der Histopathologie von LCD ebenfalls festgestellt werden (2).
LCD Typ I ist die klassische Form der LCD, die durch eine Mutation im TGFβI-Gen verursacht wird und zu einer isolierten Amyloidablagerung in der Hornhaut führt. Es wurden vier LCD-Varianten identifiziert: LCD Typ IIIA, Typ I/IIIA, Typ IV und polymorphe Amyloidose. Die LCD-Varianten treten später im Leben auf als die klassische LCD. LCD Typ IIIA tritt im 5. bis 7. Lebensjahrzehnt auf, in der Regel mit epithelialen Erosionen. Er weist dickere Gitterlinien auf, die als ropy-ähnlich“ beschrieben werden und bis zum Limbus reichen. LCD Typ I/IIIA hat dünne Gitterlinien. LCD Typ IV tritt in der 7. bis 9. Dekade mit kleinen Gitterlinien auf. Die Amyloidablagerungen bei LCD Typ IV befinden sich im tiefen Stroma, und Epithelerosionen treten selten auf. Beim polymorphen Amyloidose-Typ fehlen die Gitterlinien, und selten treten Epithel-Erosionen auf (2).
LCD Typ II ist ein systemisches Amyloidose-Syndrom, das als Meretoja-Syndrom bekannt ist und die Haut, Hirnnerven und Hornhaut betrifft. Es zeigt sich im frühen Erwachsenenalter mit peripheren Neuropathien, kranialen Neuropathien, hundeartigem Gesicht, trockener Haut, Blepharochalasis, vorspringenden Lippen und Hornhautgitterlinien. Dieser Typus wurde mit dem Gelolin-Gen auf Chromosom 9 in Verbindung gebracht, das für ein Amyloid-Vorläuferprotein kodiert, das die Aufgabe hat, Aktin an Verletzungs- und Entzündungsstellen zu entfernen (1). Der Name ist eine Fehlbezeichnung und wird nicht als Variante der gitterförmigen Hornhautdystrophie angesehen (2).
| A: Linkes Auge bei Retroillumination, das die vorderen stromalen Ablagerungen bei gitterförmiger Hornhautdystrophie zeigt | B: Linkes Auge mit höherer Leistung, das lineare vordere stromale Ablagerungen zeigt. |
 |
 |
| C: H&E-Färbung der Hornhaut mit Gitter. Man beachte die rosafarbenen amorphen Ablagerungen im Stroma | D: Eine nähere Ansicht der rosafarbenen, amorphen Ablagerungen |
 |
 |
| E: Kongorot-Färbung, die das Amyloid hervorhebt | F: Apfelgrüne Doppelbrechung des Amyloids mit Kreuzpolarisation. |
 |
 |
GRANULÄRE CORNEALE DYSTROPHIE, TYP I
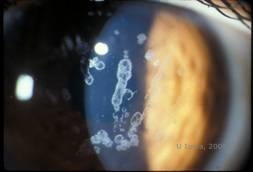
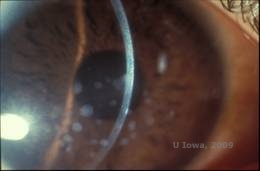
Granuläre Hornhautdystrophie, Typ I (GCD1) ist eine bilaterale, autosomal-dominante Erkrankung, die mit einer Mutation im TGFβI-Gen einhergeht und zur Ablagerung eines hyalinen Materials im Hornhautstroma führt. Sie zeigt sich typischerweise früh im ersten Lebensjahrzehnt mit grau-weißen, „krümelartigen“ Trübungen im vorderen bis mittleren Stroma, die sich bei fortgeschrittener Erkrankung bis in das hintere Stroma ausdehnen (1, 2). Diese Trübungen sind diskrete, zentral gelegene Ablagerungen mit klarer Hornhaut in der Peripherie und klarer Hornhaut zwischen den Ablagerungen (siehe Abbildung 3A und 3B). Die Krankheit ist zu Beginn typischerweise asymptomatisch, aber mit der Zeit können die Trübungen zusammenwachsen und zu einer Sehverschlechterung führen. Bei der GCD können rezidivierende Hornhauterosionen auftreten, allerdings mit geringerer Häufigkeit als bei der LCD (1, 5). Die Patienten können auch unter Blendung und Photophobie leiden (2). Zu Beginn des Krankheitsverlaufs beschränkt sich die Behandlung häufig auf Beobachtung. Wenn die Krankheit jedoch fortschreitet, können eine PTK und eine Hornhauttransplantation erforderlich sein, um das Sehvermögen zu verbessern und die Symptome zu lindern. Wie bei der LCD kann die Krankheit bei Hornhauttransplantaten wieder auftreten.
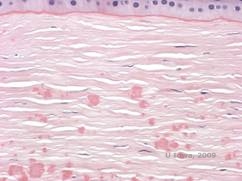
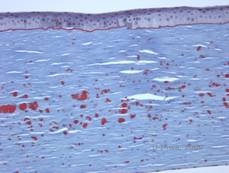
Histopathologisch handelt es sich bei den Trübungen um eosinophile Ablagerungen im vorderen Stroma, die oft als „kandisartig“ beschrieben werden und aus einem hyalinartigen Material bestehen. Mit der Zeit wandern die Ablagerungen in das tiefere Hornhautstroma. Das hyalinartige Material färbt sich mit der Masson-Trichrom-Färbung hellrot (siehe Abbildung 3C und 3D).
| A:Spaltlampenfoto der granulären Hornhautdystrophie, Typ I | B: Man beachte die „krümelartigen“ stromalen Ablagerungen mit klarem dazwischen liegendem Stroma. |
 |
 |
| C: H& E-Färbung der Hornhaut zeigt eosinophile „kandisartige“ hyaline Ablagerungen im Stroma | D: Hyalines Material färbt hellrot mit Masson-Trichrome |
 |
 |
GRANULÄRE KORNEALDYSTROPHIE, TYP II
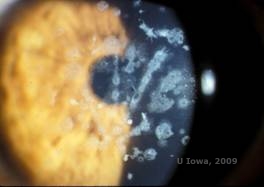
Granuläre Hornhautdystrophie, Typ II (GCD2), früher bekannt als Avellino- oder kombinierte granuläre Hornhautdystrophie, ist eine autosomal-dominante Krankheit, die mit einer Mutation im TGFβI-Gen verbunden ist und zu einer Ablagerung von Hyalin und Amyloid im Hornhautstroma führt. Typischerweise stellen sich die Patienten im zweiten Lebensjahrzehnt mit kleinen grau-weißen Punkten im oberflächlichen Stroma vor. Die Trübungen können auch dorn-, ring- oder sternförmig aussehen. Bei Retroillumination sind sie teilweise durchscheinend. Im weiteren Verlauf der Erkrankung können sich auch gitterartige Linien entwickeln (siehe Abbildung 4A und 4B). Diese Linien überkreuzen sich nicht und erscheinen weißer und weniger lichtbrechend als Gitterlinien. Die Symptome von GCD2 sind Schmerzen mit Epithelerosionen und Sehstörungen (2).
Histopathologisch weist die Hornhaut Stroma-Ablagerungen auf, die sich mit Masson Trichrom rot färben, was auf das Vorhandensein von Hyalin hinweist (siehe Abbildung 4C). Darüber hinaus zeigt eine Färbung mit Kongorot eine apfelgrüne Doppelbrechung bei Kreuzpolarisation, was auf das Vorhandensein von Amyloid hinweist (siehe Abbildung 4D). Es wurde angenommen, dass die Krankheit ihren Ursprung in einer Familie in Avellino, Italien, hat. Inzwischen wurde die GCD Typ II jedoch auch bei Patienten aus vielen anderen Ländern festgestellt (2,5), wobei die höchste Prävalenz in Ostasien besteht.
| A: Avellino-Dystrophie mit gitterartigen und körnigen Ablagerungen im Hornhautstroma | B. Masson Trichrom-Färbung mit hyalinen Ablagerungen im vorderen Stroma |
 |
 |
| C: Kongorot-Färbung mit rosafarbenen, amorphen Amyloid-Ablagerungen in derselben Hornhautprobe. | D: Kreuzpolarisation zeigt apfelgrüne Doppelbrechung, die auf Amyloid hinweist. |
 |
 |
STROMALE KORNEALE DYSTROPHIEN
MAKULARE KORNEALE DYSTROPHIE
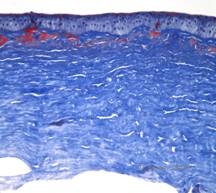
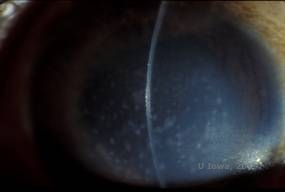
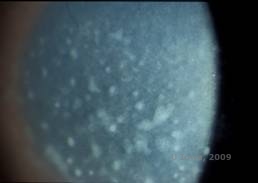
Makuläre Hornhautdystrophie (MCD) ist eine autosomal-rezessiv vererbte Krankheit, die durch eine Mutation im Kohlenhydrat-Sulfotransferase-6-Gen (CHST6) auf Chromosom 16 verursacht wird und zu einem Defekt in der Synthese von Keratansulfat führt, dem wichtigsten Glykosaminoglykan der Hornhaut. Sie ist weniger häufig als LCD oder GCD, beeinträchtigt aber das Sehvermögen stärker. Obwohl die MCD weltweit seltener vorkommt als LCD oder GCD, ist sie in Ländern wie Island und Saudi-Arabien die häufigste der stromalen Hornhautdystrophien (2,6). Grau-weiße, fleckenartige anteriore Stromaläsionen, die der GCD1 ähneln, treten in der ersten Lebensdekade in der Hornhaut auf. Im Gegensatz zu GCD1 befindet sich jedoch zwischen den Ablagerungen eine Stromatrübung, und oft ist die gesamte Hornhaut von Limbus zu Limbus betroffen (siehe Abbildung 5A und 5B). Die Hornhaut ist dünn, und mit dem Fortschreiten der Erkrankung wird die Descemet-Membran grau und entwickelt Guttae. Epithelerosionen können auftreten, sind aber bei der MCD seltener als bei der LCD. Die Patienten entwickeln in der Regel im zweiten bis dritten Lebensjahrzehnt einen schweren Sehverlust aufgrund einer diffusen Hornhauttrübung. In einigen frühen Fällen von MCD kann eine PTK durchgeführt werden. Diese Erkrankung ist jedoch im Allgemeinen nicht so gut mit PTK behandelbar wie die Gitter- oder Granulardystrophie und erfordert häufig eine Hornhauttransplantation (7). Das Wiederauftreten von Transplantaten ist bei der MCD seltener als bei der granulären oder gitterförmigen Dystrophie (1,2,5,6,8).
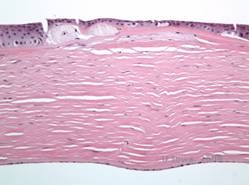
Die stromalen Ablagerungen bei der MCD bestehen aus Mucopolysacchariden, die sich im endoplasmatischen Retikulum der Keratozyten des Hornhautstromas, extrazellulär zwischen den Stromalamellen und innerhalb des Epithels, der Descemet-Membran und des Endothels ansammeln. Diese Ablagerungen färben sich mit Alcianblau blau (siehe Abbildung 5C und 5D) (1). Es gibt Brüche in der Bowman-Schicht und den Guttae mit einer Verdickung der Descemet-Membran (2).
Es wurden drei Subtypen der MCD beschrieben, die auf dem Vorhandensein oder Fehlen von immunreaktivem Keratansulfat in verschiedenen Geweben basieren. Typ I weist kein immunreaktives Keratansulfat im Hornhautstroma, in den Keratozyten, im Serum oder im Knorpel auf und ist die weltweit häufigste Variante der MCD. Beim Typ IA fehlt Keratansulfat im Stroma, in den Seren und im Knorpel, aber in den Keratozyten sind nachweisbare Mengen vorhanden. Beim Typ II ist Keratansulfat im Stroma, in den Keratozyten, in den Seren und im Knorpel in deutlich geringeren Mengen vorhanden (6).
| A: Spaltlampenfoto der makulären Hornhautdystrophie. | B: Man beachte die Trübung zwischen den stromalen Ablagerungen der Hornhaut |
 |
 |
| C: H&E der Hornhaut bei Makuladystrophie. Man beachte die vorderen stromalen Ablagerungen und die Unterbrechung der Bowman-Schicht | D: Mucopolysaccharid-Ablagerungen innerhalb der Keratozyten, hervorgehoben mit Alcianblau-Färbung |
 |
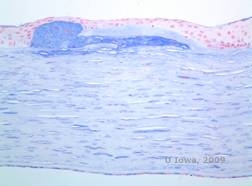 |
SCHNYDER CORNEAL DYSTROPHY (SCD)
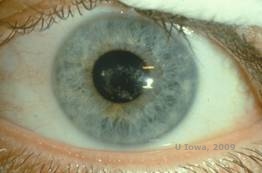
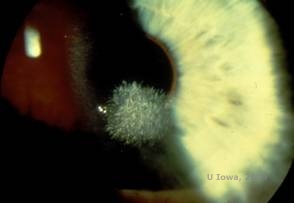
Schnyder Corneal Dystrophy (SCD), früher bekannt als Schnyder kristalline Hornhautdystrophie, ist eine autosomal dominante, bilaterale Hornhautstromadystrophie, die mit einer genetischen Mutation im UbiA prenyltransferase domain containing 1(UBIAD1) Gen auf Chromosom 1 verbunden ist. Der daraus resultierende Stoffwechseldefekt der Hornhautkeratozyten führt zu kristallinen Cholesterinablagerungen im Stroma. Das Vorhandensein von Kristallen ist jedoch für die Diagnose von SCD nicht unbedingt erforderlich. Tatsächlich weisen nur 54 % der Patienten mit SCD Hornhautkristalle auf. Typischerweise stellen sich die Patienten in der zweiten oder dritten Dekade mit einer ringförmigen zentralen Hornhauttrübung mit oder ohne kommaförmige subepitheliale Kristalle vor (siehe Abbildung 6A und 6B). Zwischen dem 23. und 38. Lebensjahr tritt dann der Arcus lipoides auf. Nach dem 38. Lebensjahr führt die fortschreitende Hornhauttrübung zu einer panstromalen Trübung, die bis in die mittlere Peripherie reicht. Bei den meisten Patienten über 50 Jahren kommt es zu einem Verlust des photopischen Sehvermögens, zu Blendungserscheinungen und einer verminderten Hornhautsensibilität, so dass eine chirurgische Behandlung einschließlich Hornhauttransplantation oder PTK erforderlich sein kann. Im Transplantat kann es zu einem Rezidiv kommen. Die Erkrankung wurde bei einigen Patienten mit Hypercholesterinämie, Hyperlipidämie und Genu valgum in Verbindung gebracht (2,5,9,10).
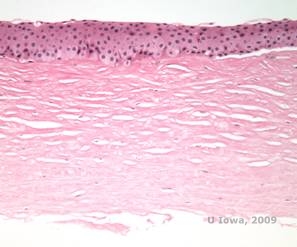
Histopathologisch lagern sich doppelbrechende Cholesterinkristalle aus Phospholipiden und Cholesterin in Basalepithelzellen, Keratozyten, der Bowman-Schicht und zwischen Stromalamellen ab. Die Lipide lösen sich bei der normalen histologischen Bearbeitung auf, so dass Gefrierschnitte durch die Hornhaut angefertigt werden müssen, um das Vorhandensein von Lipiden mit Oil-Red-O oder Sudanschwarz-Färbungen nachzuweisen.
| A: Spaltlampenfoto der Schnyder-Hornhautdystrophie. | B. Zentral gelegene kristalline Ablagerungen |
 |
 |
| C: H&E der Hornhaut mit SCCD | D. Die Öl-Rot-O-Färbung hebt Cholesterinkristalle hervor, die rot erscheinen. |
 |
 |
Tabelle 4 bietet eine gemeinsame Eselsbrücke, um sich einige der Hornhautdystrophien zu merken, die das Stroma betreffen, die Zusammensetzung ihrer Ablagerungen und die Methode der Färbung dieser Ablagerungen ist aufgeführt.
Tabelle 4: Eselsbrücke zur Erinnerung an Hornhautstroma-Dystrophien
- Marilyn-Makuläre Dystrophie
- Monroe-Mucopolysaccharid
- Immer-Alcianblau-Färbung
- Gets-Granular Dystrophie
- Her-Hyalin
- Man in-Masson Trichrom-Färbung
- Los-Lattice Dystrophie
- Angeles-Amyloid
- California-Congo Red
ÜBERSICHT: CORNEAL STROMAL DYSTROPHIEN
EPIDEMIOLOGIE
|
SIGNS
|
SYMPTOME
|
Behandlung
|
- Keefe KS, Milman T, Rodrigues MM, Hidayat AA. Conjunctival and Corneal Pathology. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3rd edition. Saunders. 2008: 3592-3595.
- Weiss JS, et al. IC3D classification of corneal dystrophies–edition 2. Cornea 2015; 34 (2): 117-159.
- Lakshminarayanan R, et al. Clinical and genetic aspects of the TGFBI-associated corneal dystrophies. Ocul Surf 2014; 12 (4): 234-251.
- Stone EM, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nature Gen. 1994; 6: 47-51.
- Bron AJ. Genetics of the Corneal Dystrophies: What we have learned in the past twenty five years. Cornea 2000; 19(5): 699-711.
- Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrierende Keratoplastik bei makulärer Hornhautdystrophie. Ophthalmology 2005;112: 220-224.
- Badr IA, Wagoner MD. Phototherapeutische Keratektomie bei makulärer Hornhautdystrophie. J Refract Surg 1999;15:481-484.
- Poulaki V, Colby K. Genetics of anterior and stromal corneal dystrophies. Seminars in Ophthalmology, 2008; 23: 1,9-17.
- Weiss JS. Schnyder corneal dystrophy. Curr Opin Ophthalmol 2009; 20 (4): 292-298
- Shearman AM, Hudson TJ, Andresen JM, et al. The gene for Schnyder’s crystalline corneal dystrophy maps to human chromosome 1p34.1p36. Hum Mol Genet 1996;5:1667-72.
Vorgeschlagenes Zitierformat: Van C, Syed NA. Epithelial-Stromal and Stromal Corneal Dystrophies: A Clinicopathologic Review. Revision of ; EyeRounds.org. August 20, 2015. Available from: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm